
The medicine man and the doctor: a virtual trip in the world of ethnopharmacology from the far west to the far east
Posted in Uncategorized on April 14, 2021 by Domenico DelfinoInvisibile
Posted in Uncategorized on February 8, 2021 by Domenico Delfino“…Non ci sono più regole. Il grande esperimento era un evento da una-volta-e-mai-più, ma ora che entrambi avete superato i vent’anni le restrizioni alla vostra follia adolescenziale non reggono più, e continuate quotidianamente ad avere rapporti sessuali per i trentaquattro giorni successivi, fino al giorno in cui parti per Parigi. Tua sorella prende la pillola, nel cassetto del suo comò ci sono creme e gelatine spermicide, hai i preservativi a disposizione, e insomma avete la certezza di essere protetti, sapete che la cosa innominabile non succederà mai: quindi potete fare l’uno all’altra tutto, e di tutto, senza timore di rovinarvi la vita. Non discutete della cosa. Al si là del breve botta e risposta della sera del compleanno di vostro fratello (Hai paura? No, non ho paura) non dite mai una parola su quello che sta succedendo, rifiutate di esplorare le diramazioni della vostra storia, del vostro mese di matrimonio, perchè questo è, alla fine, ora siete una giovane coppia di coniugi, due sposini imprigionati nei sussulti di una lussuria perpetua e prepotente – animali da sesso, amanti, amici del cuore: le ultime due persone rimaste nell’universo…”
(Tratto da Paul Auster “Invisibile”, Einaudi, 2009)
Approcci terapeutici attuali e futuri per COVID-19
Posted in Biomedical Research with tags Chang, covid-19, Nanjing University, SARS-CoV-2, terapia on January 11, 2021 by Domenico DelfinoIl coronavirus 2019 (COVID-19), classificato dall’OMS nuovo coronavirus della sindrome severa respiratoria acuta da coronavirus 2 (SARS-CoV-2), è divenuta rapidamente una pandemia globale. Per contrastare sia la sua diffusione che la virulenza, in tutto il mondo sono attive ricerche per lo sviluppo di un farmaco efficace anti-COVID-19 o un vaccino. In questa rassegna, esploriamo la comprensione clinica e la gravità dell’emergenza COVID-19 nel mondo, con un focus sulla Cina. Discuteremo inoltre i bersagli terapeutici potenziali, sia basati sul virus ospite, che potrebbero essere usati per affrontare l’epidemia COVID-19.
Introduzione
Durante le prime settimane del dicembre 2019, il primo caso di polmonite causati da un nuovo coronavirus definito come COVID-19 dal WHO è stato riportato a Wuhan, la capitale della provincia di Hubei, Cina. Si è pensato che gli impiegati locali a salario giornaliero del Huanan Seafood Market a Wuhan fossero gli iniziali portatori del COVID-19, infettando una grande proporzione della popolazione di Wuhan. Fin dalla sua emergenza e al giugno 24, 2020, COVID-19 aveva infettato circa 85098 individui in CINA. Al termine della stesura di questo articolo (24 giugno 2020), Iran, Italia, Cile, Perù, Spagna, UK, India, Russia, Brasile, e USA avevano registrato la popolazione più alta di casi COVID-19, che vanno 209970 a 2424492819 a 1537. Ad oggi, circa 9737424 casi di COVID-19 sono stati registrati in tutto il mondo, con il numero totale di morti che ha raggiunto 480140, e il numero di pazienti ricoverati di 5062840.
Nonostante le proteine codificate da SARS-CoV-2 condividano simili strutture omologhe con SARS-CoV, l’ectodominio spike (S) del virus COVD-19 mostra un’affinità di legame più alta (circa 15 nM) per la proteina recettore angiotensin-converting enzyme 2 (ACE2) del sistema bronchiale superiore, che è circa 10-20 volte più alta in paragone SARS-CoV. Quindi, questo facilita la trasmissione come mai prima di COVID-19 tra gli uomini. Il ceppo SARS-CoV-2 si può diffondere attraverso tutti i modi di contatto fisico, tra cui starnuti e tosse.
Un livello più alto di casi di COVID-19, circa l’87%, è stato registrato tra gli adulti e i gruppi di popolazione più anziana (30-79 anni). In maniera contraddittoria, un moderato (3%) o piccolo (1%) numero di casi sono stati registrati nel gruppo di popolazione più anziana (> 80 anni) e il gruppo della popolazione più giovane (10-19 anni). Concomitantemente, gente con precedenti problemi respiratori e complicazioni metaboliche, come il diabete mellito 2, ipertensione, complicazioni cardiovascolari, e cancro, sono altamente suscettibili all’infezione COVID-19 con mortalità aumentata. In accordo con evidenze cliniche precoci che gli uomini si infettano da SARS-CoV e sindrome respiratoria del Medio Oriente da Coronavirus (MERS-CoV) più delle donne, la prevalenza di COVID-19 è stata registrata nella popolazione maschile ad un ritmo più elevato in paragone alla popolazione femminile. Può darsi che l’attivazione del recettore degli estrogeni e i la associata cascata dei suoi segnali nelle donne conferisce una protezione aumentata contro l’infezione da COVID-19, similmente ai risultati di studi di ricerca basati sulla clinica con SARS-CoV e MERS-CoV.
Inoltre, alcuni pazienti con SARS-CoV-2 si sono infettati con virus da portatori asintomatici che non mostrano nessun segno clinico ovvio, come influenza, stanchezza, febbre, e tosse secca, ma sono capaci di trasmettere il COVID-19 a individui sani con contatti fisici diretti o indiretti attraverso goccioline nasali e starnuti. In aggiunta, a questa principale barriera al controllo della diffusione di COVID-19, è difficile controllare la diffusione della malattia da pazienti guariti ad individui sani. In questa rassegna, ci focalizzeremo su studi clinici che coinvolgono farmaci contro COVID-19, bersagli terapeutici clinici potenziali, e direzioni future della gestione di COVID-19.
Insegnamenti clinici appresi, attuali molecole terapeutiche, e prospettive nella gestione del COVID-19
Come illustrato nella figura 1, tutte le proteine non strutturali (NPS), che rappresentano i bersagli basati sul virus, sono cruciali per progettare gli sforzi per migliorare la colonizzazione del virus nell’ospite, soprattutto nell’alto sistema bronchiale. I siti catalitici delle NPS funzionali dei coronaviridiae possono essere presi di mira per attenuare la virulenza di SARS-CoV, MERS-CoV, e SARS-CoV-2. Inoltre, le NPS funzionali interagiscono con ACE2 dell’ospite per consentire la penetrazione del coronavirus nella cellula.
Vista la mancanza attuale di un vaccino che possa attenuare o prevenire la trasmissione di COVD-19, prendere di mira qualsiasi dei momenti cruciali di invasione di SARS-CoV-2, come la penetrazione del virus, trascrizione e traduzione, sintesi del genoma ed assemblaggio, e rilascio del virus dovrebbe essere efficace nell’invertire la sua patogenesi e trasmissione nell’uomo.
Per prendere di mira la colonizzazione iniziale di SARS-CoV-2, la ricerca si è focalizzata sull’uso di potenziali agenti antivirali contro altri virus, come SARS-CoV, MERS-CoV, epatite B, epatite C, HIV, e virus comune dell’influenza.
Lezioni dal passato
Durante le fasi iniziali dell’epidemia COVID-19, dal tardo dicembre 2019 ai primi di febbraio 2020, mentre sono stati confermati 40.000 casi e 850 persone sono morte in tutta la Cina, i pazienti in studio clinico sono stati trattati con farmaci che si sono rivelati generalmente inefficaci contro il virus. Uno di questi farmaci era oseltamivir, selezionato in quanto i sintomi COVID-19 includevano febbre e altri sintomi comuni nelle infezioni influenzali. Oseltamivir è un potente inibitore della neuraminidasi che attenua efficacemente la virulenza dei virus dell’influenza A e B, ma non ha mostrato alcun effetto evidenziabile contro COVID-19 in quanto il virus non secerne neuraminidasi. Inoltre, il trattamento di pazienti COVID-19 con farmaci antibatterici, tra cui moxifloxacina, ceftriaxone, e azitromicina, sia singolarmente che in associazione, ha mostrato solo piccoli benefici. In aggiunta, è stato documentato che l’uso a lungo termine di dosi elevate di antibiotici durante l’epidemia da COVID-19 era legato con sintomi clinici avversi di gravi disturbi respiratori, inclusi iperinfiammazione, shock, alterazione circolatoria, e danni ad altri organi.
I corticosteroidi che mimano i corticosteroidi naturali del corpo umano sono stati frequentemente usati per trattare pazienti con ghiandole surrenali deficienti che non erano in grado di sintetizzare livelli sufficienti di corticosteroidi. Tuttavia, i corticosteroidi sono anche stati ampiamente prescritti a dosi elevate in pazienti con disordini immunologici, infiammazione, e/o alterato equilibrio idro-salino. Nonostante ciò, non ci sono chiare evidenze di benefici terapeutici fondamentali dei corticosteroidi nel trattamento dei sintomi respiratori del virus respiratorio sinciziale (RSV), dei virus dell’influenza comune, SARS-CoV, e MERS-CoV. Ricerche sono state condotte per valutare l’efficacia dei corticosteroidi in dosi da basse a medie per trattare i sintomi clinici avversi indotti dai coronavirus. Allo stesso tempo, i pazienti con COVID-19 sono stati trattati con corticosteroidi come farmaco supplementare per una durata minima di 3-15 giorni, senza sostanziali miglioramenti. Questo tempo più breve di trattamento è stato attuato a causa dei ben noti effetti avversi a lungo termine e altri livelli secondari di complicazioni.
Quindi, la mancanza di successo con i citati farmaci hanno anche contribuito all’aumento di mortalità tra i pazienti con COVID-10 in tutta la Cina, insieme all’alterazione nella diagnosi precoce e al trattamento di COVID-19; l’azione ritardata da parte degli operatori sanitari per interrompere la catena di trasmissione del COVID-19, una scarsa comprensione della virulenza del COVID-19 e della sua efficace trasmissione nella popolazione comune; e inadeguati kit di diagnosi clinica e di altre strumenti medici essenziali, come ventilatori respiratori, camici e guanti medici protettivi, per gestire pazienti criticamente affetti da COVID-19.
Terapeutici attuali con significato clinico per la gestione di COVID-19
Agenti chimici
Per gestire l’epidemia emergente di COVID-19 e la sua associata mortalità, circa 300 studi clinici sono stati eseguiti su pazienti con COVID-10 in Cina. Alcuni dei farmaci clinici usati in questi studi hanno mostrato risultati promettenti nel contrastare i sintomi clinici di COVID-19 (Tabella 1).
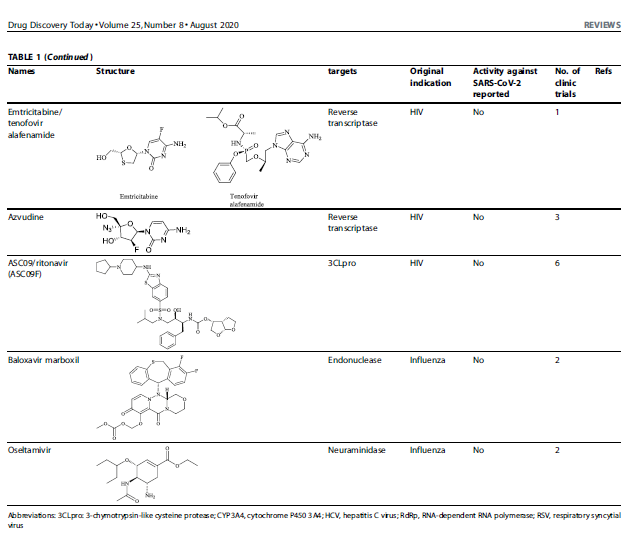
Per combattere l’epidemia SARS-CoV, la combinazione farmacologica lopinavir e ritonavir [approvata dalla US food and drug administration (FDA)] che inibiscono la proteasi virale 3-chymotrypsin-like cysteine, addizionata di ribavirin, ha efficacemente controllato la virulenza di SARS-CoV e il ritmo di mortalità associato ad esso. Questi effetti sono risultati in circa 14 studi clinici che hanno usato la combinazione farmacologica lopinavir e ritonavir per trattare pazienti con COVID-19. La combinazione farmacologica di lopinavir e ritonavir che ha efficacemente attenuato i sintomi clinici avversi, come la febbre, di cinque pazienti con COVID-19 richiede ulteriori validazioni cliniche. Al contrario, un recente studio clinico che ha usato questa combinazione farmacologica non ha mostrato nessun evidente effetto terapeutico su pazienti adulti con COVID-19 rispetto ad altri pazienti che avevano ricevuto medicazioni classiche. Quindi, c’è bisogno di ulteriori approfonditi studi clinici su questa combinazione di farmaci.
Ribavirin, un altro farmaco antivirale approvato dall’FDA comunemente usato per trattare il virus dell’epatite C e RSV, è stato ampiamente suggerito in combinazione con antibiotici efficaci e/o con o senza trattamento ormonale. Come potente inibitore dell’RNA polimerasi virale RNA-dipendente (RdRp), la ribavirina ha efficacemente controllato la virulenza di COVID-19 quando era co-somministrata con la combinazione farmacologica ritonavir/lopinavir.
Dato che i derivati della clorochina esibivano una forte azione inibitrice contro la colonizzazione SARS-CoV, i clinici hanno mostrato che i derivati della clorochina insieme a remdesivir controllava efficacemente la proliferazione di ceppi clinicamente isolati di SARS-CoV-2. Uno studio clinico con clorochina fosfato per trattare la virulenza di COVID-19 è stato approvato dalla Commissione Nazionale della Salute della Repubblica Popolare Cinese.
Arbidol, un potente farmaco antivirale che ha come bersaglio citochine infiammatorie associate al virus, è stato usato per trattare pazienti con COVID-19 e polmonite grave; Non ha mostrato alcun effetto avverso e, quindi, è in studio sia in Cina che in Russia. In aggiunta, studi sia in vivo che in vitro hanno esplorato gli ulteriori vantaggi terapeutici dell’arbidol come farmaco che esibisce una forte risposta immune rompendo il capside virale legato alla membrana delle cellule ospiti. Arbidol è stato ritenuto prioritario insieme agli altri farmaci clinici potenziali discussi prima per studi clinici di contrasto all’epidemia da COVID-19, supportati dalla sesta edizione delle Guidelines for the Prevention, Diagnosis, and Treatment of Novel Coronavirus-induced Pneumonia.
E’ stato suggerito che il dipiridamolo, un farmaco antipiastrinico e inibitore della fosfodiesterasi che ha come bersagli i livelli intracellulari di cAMP/cGMP, inclusi i virus a filamento positivo di RNA, possa essere un efficace farmaco antivirale. Il dipiridamolo inibisce efficacemente la replicazione di SARS-CoV alla concentrazione efficace 50 (EC50) di 100 nM in vitro. Questa capacità clinica del dipiridamolo enfatizza il suo possibile utilizzo come adiuvante per rinforzare il sistema immune così come per inibire la proliferazione virale e l’ipercoagulazione.
Darunavir, un potente inibitore retrovirale, insieme a cobicistat, che controlla l’attività del citocromo P4503A (CYP3A) determinando la rottura degli agenti antivirali, è stato usato per trattare pazienti con HIV. Nonostante Darunavir sia inteso per inibire le proteinasi virali, sono necessarie ulteriori ricerche per provare il suo significato clinico nel contrastare la virulenza di COVID-19.
Studi animali hanno mostrato che remdesivir inibisce la virulenza di SARS-CoV e MERS-CoV. Anche ricerche in vitro hanno confermato che il trattamento con remdesivir inibiva profondamente la proliferazione di SARS-CoV-2 ad un EC50: 0.77-1.76 μM.
Favipiravir, che inibisce marcatamente l’RNA polimerasi dipendente dall’influenza, esibisce una profonda attività antivirale contro molti virus, tra cui arenavirus, bunyavirus e filovirus, che determinano febbre emorragica fatale. In aggiunta a questi virus, il trattamento con favipiravir inibisce efficacemente la proliferazionedi SARS-CoV-2 in colture di cellule Vero E6, con un valore di EC50 di 62 μM. In base a questi esiti positivi, il Ministro di Scienza e Tecnologia, Cina ha recentemente raccomandato il favipiravir per la gestione del COVID-19 in un gruppo più grande di pazienti.
Altri farmaci potenziali con elevato valore terapeutico includono baloxavir e marboxil, che hanno come bersaglio l’endonucleasi virale dipendente dal cap (anche se questa è assente in SARS-CoV-2); TMC310911 ha come bersaglio l’attività proteasica virale; emtricitabina/tenofovir alafenamide e azvudina hanno come bersaglio la trascrittasi inversa virale e sono stati testati in pazienti con COVID-19. Nonostante questi farmaci clinici abbiano mostrato effetti terapeutici promettenti contro la virulenza di COVID-19, c’è ancora bisogno di uno studio clinico più profondo per confermare la loro efficacia in un gruppo di pazienti con COVID-10 più grande.
La farmacocinetica (PK) di tutti i farmaci emergenti COVID-19, come assorbimento, distribuzione, metabolismo ed escrezione, inclusi gli effetti farmacologici sull’organismo ospite (farmacodinamics; PD) , possono essere studiati usando modelli PK/PD. Un modello farmacologico PK/PD rivelerebbe la correlazione tra esposizione al farmaco e i duoi effetti PD associati in silico. Dei modelli empirici e meccanicisti disponibili di PK/PD, i modelli empirici che comprendono un link diretto, funzioni chiavetta, regressione logistica, e modelli circadiani potrebbero essere usati per studiare la progressione della malattia in pazienti con COVID-19 con o senza esposizione in silico ai farmaci. Studi di modelli meccanicistici, che si basano principalmente su dati di biomarcatori clinici nel contesto della virulenza di COVID-19 in presenza o assenza di farmaci, conferirebbero una breve comprensione clinica dell’effetto farmacologico contro COVID-19 nell’uomo e in altre specie così come il dosaggio farmacologico determinato per la gestione di COVID-19. Le analisi PK/PD sono condotte a tre diversi livelli: il livello 1 rivela la correlazione diretta tra l’esposizione al farmaco e la sua risposta rilevante usando un plot grafico misurando la concentrazione plasmatica del farmaco libero graficata contro la risposta PD rilevante (in vivo); ciò genera la concentrazione efficace del dosaggio farmacologico in base al rapporto tra la media della concentrazione plasmatica del farmaco libero/concentrazione inibitoria al 50% (IC50); il livello 2 genera il tasso di ricambio della risposta PD, Kout , in risposta ai cambiamenti misurabili nel farmaco e nel sistema biologico; il livello 3 (con i modello di supporto prestabiliti) chiarisce la risposta farmacologica al farmaco a dosaggi vari tra i pazienti coinvolti nello studio sperimentale. La correlazione dei biomarcatori con i modelli PD/PK generati fornirebbero anche una comprensione meccanicistica del meccanismo d’azione dei farmaci che possa consentire la ricerca traslazionale e la valutazione intersoggetto del farmaco su scala più ampia.

Agenti biologici
La terapia con plasma di convalescenti usando campioni di plasma sanguigno procurati clinicamente di pazienti con un determinato virus sono stati adottati per trattare pazienti con SARS-CoV-2. E’ stato proposto che l’infusione di plasma sanguigno di convalescenti (CBP) in questi pazienti dovrebbe efficacemente attenuare la patogenicità del virus, con la sua eventuale rimozione dal sangue del paziente. Nonostante la metodologia di trasfusione CBP abbia alcune costrizioni per gli studi clinici, il trattamento clinico usando CBP di pazienti con COVID-19 è stato considerato sia promettente che efficace per trattare pazienti criticamente affetti da COVID-19. Per saggiare la trasfusione CBP su larga scala, alcuni fattori clinici devono essere presi in considerazione, come la trasmissione di patogeni intermittenti tra il donatore ed il recipiente e il preciso reclutamento dei donatori con un sufficiente titolo anticorpale per produrre un effetto evidente contro la patogenicità di quel particolare patogeno nei pazienti.
Cellule staminali mesenchimali derivate dal midollo osseo (MSC) sono state usate per trattare pazienti con sindrome da distress respiratorio acuto (ARDS), senza effetti avversi registrati durante lo studio rilevante. Al momento, ci sono 13 studi clinici in esecuzione per gestire SARS-CoV-2 usando MSC, con risultati clinici iniziali promettenti in sette pazienti con COVID-19, che hanno mostrato un rimarchevole miglioramento delle loro condizioni cliniche entro 14 giorni di trattamento senza nessun evidente effetto avverso.
L’Interferon-alpha (IFN-α), una potente citochina immune rilasciata durante l’infezione da patogeni, ha migliorato la funzione polmonare quando veniva accoppiato con altri agenti anti-virali, come il lopinavir, ritonavir, e remdesivir, in pazienti con MERS-CoV. La combinazione terapeutica di ribavirin e IFN-α è stata riconosciuta nella sesta edizione delle Guidlines for the Prevention, Diagnosis, and Treatment of Novel Coronavirus-induced Pneumonia.
Pazienti con COVID-19 ed altre gravi condizioni di salute hanno mostrato livelli circolatori aumentati di citochine proinfiammatorie, soprattutto interleuchina-6 (IL-6), che potrebbe essere responsabile dei sintomi clinici avvfersi, come shock settico, danno ai tessuti d’organo associati a cuore, fegato, e rene, e disfunzione respiratoria. In uno studio clinico in esecuzione (ChiCTR2000029765), i clinici hanno preso come bersaglio gli aumentati livelli di IL-6 usando un anticorpo monoclonale tocilizumab specifico per IL-6 per trattare pazienti criticamente affetti con COVID-19. I risultati clinici di 21 pazienti Cinesi con COVID-19 hanno dimostrato una riduzione della temperatura corporea dopo il trattamento con miglioramento della loro funzione respiratoria. Questi risultati suggeriscono di avere come bersaglio anche altre citochine proinfiammatorie circolatorie, come IL-1 e IL-17, usando anticorpi neutralizzanti specifici per le citochine. In seguito al successo nei risultati di questi studi clinici, dovrebbe essere possibile adottare una strategia biologica proficiente per trattare la virulenza del COVID-19 in pazienti immunocompromessi. Un anticorpo monoclonale etichettato “CR3022”, generato contro il dominio di legame al recettore della glicoproteina S di membrana di SARS-CoV-2, potrebbe essere di beneficio ai pazienti interrompendo la sua colonizzazione nel tratto alto del sistema respiratorio. Inoltre, anticorpi monoclonali generati specificamente contro le proteine funzionali di SARS-CoV-2 e/o il suo agonista potenziale ACE2, che controlla la penetrazione virale, dovrebbe dare un significativo beneficio ai pazienti in termini di un rapido recupero e un ristretto tasso di trasmissione.

Strategie cliniche future per la gestione di COVID-19
Per contrastare l’epidemia da COVID-19 all’interno di una breve cornice, trattamenti che usano farmaci riproposti contro bersagli basati sul virus e sull’ospite potrebbero risolvere problemi clinici simili in futuro. Nel lungo termine, lo sviluppo di farmaci antivirali nuovi-a molte faccie-pan CoV contro i coronavirus potrebbero risultare in un trattamento efficace per SARS-CoV-2. Una profonda attivazione dei recettori bitter taste [taste 2 receptor member 4 (T2R4); taste 2 receptor member 38 (T2R38); taste 2 receptor member 43 (T2R43) e taste 2 receptor member 46 (T2R46)] usando composti bitter taste, tra cui nicotina (come agonista), potrebbe attenuare la virulenza di COVID-19 con l’aumento della produzione di ossido nitrico (NO)intracellulare calcio-dipendente accompagnata dalla ridotta secrezione di citochine proinfiammatorie nel sistema respiratorio alto. Questo aumento nella produzione di NO rafforza ulteriormente la frequenza del battito ciliare con risultante pulizia mucociliare dei patogeni invasori. L’idea di stimolare la risposta immune innata tramite l’attivazione di recettori bitter taste, e la riduzione della produzione di citochine proinfiammatorie tramite stimolazione di ACE2 e i recettori neuronali dell’acetilcolina usando nicotina, saranno validati in pazienti COVID-19 nel prossimo futuro.
Considerazioni conclusive
Tutte le strategie terapeutiche proposte e discusse fin qui, inclusi gli studi clinici in corso per la gestione del COVID-19, devono superare ostacoli sostanziali, come la possibilità di mutazioni spontanee di SARS-CoV-2, modelli animali ristretti per studi preclinici, una mancanza di pazienti per gli studi clinici, L’alto costo di mantenimento degli scenari sperimentali, e la ritenzione della sostenibilità dei risultati degli studi terapeutici basati sulla clinica. In aggiunta, tutti i criteri di base di ciascuno studio clinico devono essere ben studiati ed obbedire alle linee guida cliniche appropriate, indipendentemente dal tipo di studio. Solo con questi sforzi concertati di ricerca è probabile che la comunità scientifica abbia successo nella ricerca per un terapeutico con provati effetti contro il COVID-19.
(Tratto da Yongtao Duan, Yongfang Yao, Senthil Arun Kumar, Hai-Liang Zhu, Junbiao Chang. “Current and future therapeutical approaches for COVID-19”. Drug Discov Today, 2020 Aug;25(8):1545-1552)
Il gene egoista
Posted in libri with tags DNA, gene egoista, Richard Dawkins, virus on October 19, 2020 by Domenico Delfino“…all’interno di tutte le nostre cellule ci sono numerosi organelli chiamati mitocondri. I mitocondri sono fabbriche chimiche, responsabili delle forniture di energia di cui abbiamo bisogno. Se perdessimo i nostri mitocondri moriremmo in pochi secondi. Recentemente si è sostenuto in modo plausibile che i mitocondri erano in origine batteri simbionti che molto tempo fa si sono uniti con il nostro tipo di cellule. Suggerimenti simili sono stati fatti per altri organelli che si trovano all’interno delle cellule. Questa è una delle idee rivoluzionarie che richiedono tempo per essere accettate, ma il momento ormai è giunto. Io penso che arriveremo ad accettare l’idea ancor più radicale che ciascuno dei nostri geni è un’unità simbiotica. Siamo colonie gigantesche di geni simbiotici. Non si può parlare di vere e proprie “prove” di questa idea ma, come ho cercato di suggerire nei capitoli precedenti, queste sono in effetti inerenti al modo stesso in cui pensiamo al funzionamento dei geni nelle specie sessuate. L’altra faccia della medaglia è che i virus possono essere geni evasi da “colonie” simili a noi. I virus consistono di DNA puro (o di una molecola simile che si autoreplica) circondato da un rivestimento proteico e sono tutti parassiti. L’idea è che si siano evoluti da geni “ribelli” che sono fuggiti e che ora viaggiano da un corpo all’altro direttamente attraverso l’aria, invece che per mezzo di veicoli più convenzionali come gli spermatozoi o le cellule uovo. Se questo è vero, potremmo considerare anche noi stessi come colonie di virus! Alcuni di essi cooperano in modo simbiotico e viaggiano da un corpo all’altro tramite spermatozoi e cellule uovo: sono i “geni” convenzionali. Altri vivono da parassiti e viaggiano con qualunque mezzo possibile. Se il DNA parassita viaggia tramite spermatozoi e cellule uovo, forse forma quel surplus “paradossale” di DNA che ho menzionato nel capitolo 3; se viaggia attraverso l’aria o con altri mezzi diretti si chiama “virus” nel senso comune della parola….”
(Tratto da Richard Dawkins, “Il gene egoista – la parte immortale di ogni essere vivente”, Scienza Oscar saggi Mondadori, 1992)
Il fiume mare
Posted in Viaggi with tags fiume, Montreal, Paolo Galliani, Québec, San Lorenzo on July 24, 2020 by Domenico Delfino“…”A Montreal, nemmeno negli asili avrebbero altrettanta cura dei bambini”, dicono da queste parti. Possibile, anzi certo. Più ancora dei cetacei, sono loro – i biologi – il grande miracolo di questo pezzo di Québec: si finisce per ascoltare a bocca aperta Chantal St-Hilaire mentre spiega le differenze tra i 13 tipi di balene che frequentano queste acque, per invidiare Nadia Ménard che studia la biodiversità del parco marino Saguenay-Saint-Laurent e per sorridere quando Robert o Veronik del Gremm (Groupe de recherche et l’Education sur les Mammiféres Marins) analizzano il materiale fotografico sulle megattere di Tadoussac con la stessa tenerezza dei nonni e degli zii mentre sfogliano gli “album di famiglia” con le immagini dei nipotini…
La Nazionale 308 che arriva a Québec si blocca davanti al fiordo di Sagunay e l’attesa del traghetto che va e viene da Tadoussac regala la vista sorprendente di quattro beluga che danzano nell’acqua a pochi metri dalla riva in una sorta di nuoto sincronizzato. “Ne conosciamo almeno 450”, spiega Patrice Corbeil, direttore dell’istituto di rue de la Cale Sèche, “li seguiamo, valutiamo il loro stato di salute, periodicamente effetuiamo anche delle bipsie perché purtroppo cercano cibo nei sedimenti impregnati di sostanze contaminanti e tossiche. Quando serve li rimorchiamo presso una facoltà vicina di veterinaria per le cure antitumorali”. E il suo entusiasmo fa rima con quello del quotidiano locale Le Soleil che titola: “Les bailenes revienent”. Le balene stanno tornando da queste parti più di quanto sia mai successo negli ultimi 17 anni. Devono essere le norme più severe per ridurre lo scarico di inquinanti che arrivano dal mondo agricolo o forse sono le navi in transito che rispettano le regole sulla velocità moderata nelle aree fluviali frequentate dai cetacei.
Il ferry è intanto arrivato ed è davvero un attimo passare sulla sponda orientale del fiordo, frontiera fisica ma anche simbolica fra la zona più urbanizzata e quella più disabitata della Costa Nord: da qui in poi è un altro mondo. Dodici miseri minuti eppure c’è gente che vorrebbe azzerarli costruendoci sopra un viadotto. Scusa pronta: favorirebbe lo sviluppo economico delle regioni più remote. Come se fosse facile piazzare una sorta di ponte di Brooklyn proprio qui, davanti a questo San Lorenzo che due volte all’anno si tinge di bianco, quando decine di migliaia di oche delle nevi vengono a planare, facendo impazzire gli ornitologi e divertire i bambini. Al Gremm i biologi hanno finito di completare la scheda di Witche’s Hat, una balena che guida un grosso branco. Ma sulle loro scrivanie ci sono le foto anche di altri cetacei, di Donna Vitale, di Dacks, di Hang Nail. C’è il giovane Chap Chap e c’è pure Ratatouille che fa i capricci. Chi l’ha detto che gli scienziati non possano essere anche dei poeti… ”

Da sinistra, la chiesetta della comunità Innus di Pointe-Parente e quella di Tadoussac con vista sul fiume (foto di Bruno Zanzottera/Parallelo Zero).
(Tratto da Paolo Galliani “Il Fiume Mare”, Meridiani Québec, Editoriale Domus, maggio 2008, n. 168)
Mistici e maghi del Tibet
Posted in libri with tags Alessandra David-Neel, Amdo, lama, Thags-Yang, Tibet on June 17, 2020 by Domenico Delfino“…Un giovane che conosco fu mandato dal suo maestro – un lama di Amdo – in un burrone solitario, assai tetro, che passava per essere frequentato dagli esseri malvagi. Egli doveva attaccarvisi contro una roccia; poi, venuta la notte, evocare e sfidare le deità sanguinarie più feroci, quelle che i pittori tibetani dipingono mentre succhiano il cervello degli uomini e svuotano gli intestini.
Per quanto grande potesse essere il suo terrore, gli era stato ordinato di resistere alla volontà di slegarsi per fuggire e di rimanere allo stesso posto fin dopo il levar del sole.
Questa pratica è, per così dire, classica e serve di debutto a molti novizi tibetani sul sentiero mistico.
Talora, è comandato al discepolo di rimanere attaccato durante tre giorni e tre notti, od anche di più, a digiuno, lottando contro il sonno, in preda alla fame ed alla stanchezza che generano così facilmente delle allucinazioni.
Il risultato tragico di un esercizio di questo genere fu raccontato a Yongden da un vecchio lama di Tsarong, durante il mio viaggio in incognito a Lhassa. E’ ben inteso che, seduta in un angolo, la “trascurabile mamma” che figuravo allora, non perdette una parola del racconto.
Nella loro gioventù, questo lama e suo fratello minore, chiamato Lodeu, avevano lasciato il loro monastero per attaccarsi ad un asceta straniero alla regione, che si era temporaneamente stabilito come eremita su una montagna, ben conosciuta come luogo di pellegrinaggio, chiamata Phagri, non lontano da Dayul.
L’anacoreta comandò al più giovane dei due fratelli di andare ad attaccarsi per il collo ad un albero, in un luogo frequentato, si diceva, da Thags-yang, un demonio che appare generalmente sotto la forma di una tigre ed al quale si attribuiscono gli istinti feroci di questa belva.
Così legato, come una vittima al palo del sacrificio, l’uomo doveva immaginarsi di essere una vacca condotta in quel luogo come offerta propiziatrice a Thags-Yang. Fissando tutti i suoi pensieri su quest’idea e mugghiando, di quando in quando per entrar meglio nella parte che rappresentava, egli aspetterebbe – se la sua concentrazione di pensiero fosse abbastanza potente – uno stato di transe nel quale avendo perduto la coscienza della sua personalità, si sentirebbe una mucca in pericolo di essere divorata.
L’esercizio doveva essere continuato tre giorni e tre notti consecutivi.
Passarono quattro giorni senza che il novizio ritornasse dal suo maestro. Il mattino del quinto giorno, questo disse a quello dei due discepoli che era rimasto con lui:
– La notte scorsa ho fatto uno strano sogno, Andate a prendere vostro fratello.
Il monaco partì per il luogo in cui questo era stato mandato.
Lo attendeva uno spettacolo orribile: il cadavere di Lodeu, straziato e mezzo divorato era ancora là, in parte attaccato ad un albero, mentre brani sanguinolenti giacevano fra i cespugli circostanti.
Impressionatissimo, l’uomo raccolse i resti funebri nella sua toga monastica e si affrettò a ritornare verso il suo maestro.
Quando arrivò alla capanna che serviva di ricovero all’eremita ed ai suoi discepoli, la trovò vuota. Il lama era partito, portando via con se tutta la sua sostanza: due libri religiosi, alcuni oggetti rituali ed il suo bastone da viaggio sormontato da un tridente.
– Mi sentivo impazzire, – racconta il vecchio tibetano. – Ancor più che la scoperta del corpo di mio fratello, questa inspiegabile partenza mi spaventava. Che cosa aveva sognato il nostro maestro? Conosceva egli forse la triste sorte del suo discepolo. Perchè era sparito?…
Senza poter meglio indovinare che il povero monaco le ragioni che avevano indotto il lama a fuggire, immaginai tuttavia, che non vedendo ritornare il suo allievo, egli avesse potuto sospettare un accidente del genere di quello che realmente era accaduto. Forse egli aveva avuto, veramente, uno di quegli avvertimenti misteriosi che talora portano i sogni ed aveva prudentemente evitato la collera dei parenti della vittima.
In quanto alla morte del giovane, essa poteva spiegarsi in modo naturale. Le pantere sono numerose in quella regione; anche i leopardi vi scorazzano. Ne avevo incontrato nella foresta, pochi giorni prima di udire questa storia. Uno di essi, che il novizio aveva forse egli stesso attirato coi suoi muggiti, ne aveva fatto la sua preda prima che avesse avuto il tempo di slegarsi per tentare di difendersi.
Però, l’opinione del monaco che narrava la storia e di coloro che lo circondavano era differente. Per essi, lo stesso demonio-tigre si era impossessato dell’offerta imprudentemente esibita. Il giovane discepolo, essi dicevano, ignorava probabilmente le parole ed i gesti magici che l’avrebbero protetto. Ed in questo il suo maestro era colpevole di avergli fatto affrontare la presenza del “demonio-tigre” senza averlo armato cogli insegnamenti dell’indispensabile iniziazione.
Tuttavia, in fondo alla sua mente, il fratello, ferito nella sua affezione, conservava un pensiero più terribile che esprimeva a bassa voce, tremando.
– Chi sa, – egli diceva, – se questo lama straniero non era egli stesso il demonio-tigre, metamorfosato in uomo per attirare una vittima. Egli non poteva impadronirsene sotto la sua apparenza umana e la notte, mentre dormiva, riprendendo la sua forma di tigre, egli aveva soddisfatto la sua ferocia.
Un silenzio profondo succedette alle parole del vecchio. Egli aveva dovuto raccontare sovente questo terribile episodio della sua lontana giovinezza, ma la storia non aveva cessato di impressionare i suoi uditori.
No era forse sempre di attualità? Thags-Yang e tanti altri della sua specie non continuavano forse a gironzolare attorno agli uomini ed agli animali insufficientemente protetti contro le loro imprese?
Nella grande cucina soltanto rischiarata dalle fiamme del focolare, una ventata di angoscia passava sulla famiglia radunata. Una donna alzava istintivamente gli occhi verso i fogli di carta incollati sul muro e portanti i segni magici protettori, come per constatare che erano sempre là. Il nonno andò ad assicurarsi che la lampada dell’offerta vespertina era accesa sull’altare, nella stanza vicina e l’odore dei bastoni di incenso che egli accendeva si diffuse nella casa…”
(Tratto da Alessandra David-Neel, “Mistici e maghi del Tibet”, Milano, Fratelli Bocca Editori, 1949)
Rompere le barriere allo sviluppo di nuovi farmaci analgesici
Posted in Farmaci in Anestesiologia with tags abuso, analgesici, canali ionici, clifford J Woolf, dolore cronico on June 17, 2020 by Domenico DelfinoRiassunto
Problemi di dolore acuto e cronico, ancorchè molto frequenti, sono in genere poco soddisfatte dalle terapie esistenti. I bisogni clinici irrisolti riflettono il fallimento nello sviluppo di nuove classi di analgesici con efficacia superiore, con diminuiti effetti avversi e una più bassa tendenza all’abuso rispetto a quelli attualmente disponibili. I motivi di ciò includono l’eterogeneità delle condizioni cliniche di dolore, la complessità e diversità dei sottostanti meccanismi patofisiologici accoppiate all’inattendibilità di alcuni modelli preclinici di dolore. Tuttavia, recenti avanzamenti nella comprensione della neurobiologia del dolore hanno cominciato ad offrire opportunità allo sviluppo di nuove strategie terapeutiche e alla rivisitazione di bersagli esistenti, tra cui la modulazione di canali ionici, di enzimi e GPCR.
Introduzione
Il dolore è la ragione principale della ricerca di cure mediche da parte dei pazienti, con oltre il 40% della popolazione US afflitta da dolore cronico. Solo negli US nel 2013, il costo totale per il trattamento di certe condizioni di dolore cronico è ammontato a > 130 miliardi di $. Gli analgesici disponibili – FANS, inibitori della ricaptazione delle amine, farmaci antiepilettici ed oppioidi – hanno efficacia analgesica variabile ma tipicamente di basso livello, e sono in genere associati ad effetti deleteri. Infatti, gli oppioidi, che sono quelli più frequentemente usati (240 milioni circa di prescrizioni nel 2014) e spesso la classe più efficace di analgesici, producono tolleranza, dipendenza, e stipsi, e sono associati con tendenze all’abuso maggiore, mentre la depressione respiratoria associata a dosi elevate ha portato ad un aumento catastrofico nel numero di morti per overdose (OD) a queste sostanze nel nord America.
Diverse situazioni patologiche in diversi siti anatomici possono determinare dolore. Le cause di dolore includono cancro, infiammazione o danno tessutale, così come danni o lesioni del sistema nervoso. Possono anche insorgere diverse sindromi di dolore cronico diffuso a causa dell’anormale stato di amplificazione all’interno del CNS. Tutto ciò può portare, attraverso l’attività anormale dei sistemi nocicettivi, a dolore in assenza di uno stimolo (dolore spontaneo), risposte esagerate a stimoli nocivi (iperalgesia) e dolore evocato da stimoli normalmente innocui (allodinia).
Data la prevalenza della diversione ed overdose oppioidea, c’è chiaramente un bisogno urgente di sviluppare nuovi analgesici efficaci e sicuri senza tendenza all’abuso. Il lavoro di un grande numero di laboratori ha gettato nuova luce sui meccanismi e le vie che mediano aspetti specifici dei nessi nocicettivi (fig. 1), rivelando il potenziale per altri bersagli fini del dolore acuto e cronico in diverse condizioni cliniche. Stanno emergendo strategie terapeutiche completamente nuove che hanno come bersagli canali ionici, enzimi e GPCR, spesso con una validazione genetica umana. In questa rassegna, presentiamo questi meccanismi emergenti, e valutiamo nuovi bersagli ed agenti negli stadi preclinici e clinici precoci di sviluppo che mostrano promesse per lo sviluppo di analgesici innovativi (Tabella 1 e 2).
L’eterogeneità delle condizioni di dolore clinico e la complessità e molteplicità dei sottostanti meccanismi patofisiologici ha reso difficile identificare bersagli trattabili con ampio coinvolgimento – il grande modello di un trattamento per tutte le condizioni di dolore non è sostenibile. La scarsa prevedibilità dei modelli preclinici di “dolore” può risultare nella selezione di candidati che non possiedono attività nelle condizioni sofferte dai pazienti (Box 1).
Al contrario, le difficoltà nell’assicurare la stimolazione del bersaglio, la perdita di sensibilità degli studi clinici, e le distorsioni indotte dal placebo aumentano il rischio che composti o bersagli potenzialmente efficaci possano essere abbandonati prematuramente. Questi aspetti hanno portato ad indirizzare i maggiori sforzi di sviluppo a riformulazioni di classi già validate di analgesici esistenti: oppioidi, FANS, agenti anti-epilettici ed inibitori della captazione di amine, nonostante i loro ben noti limiti.
______________________________________________________________________________________________
Box 1
Le sfide dei modelli preclinici di dolore
Modelli preclinici efficaci nei roditori sono essenziali per lo sviluppo di analgesici, ma la loro validità predittiva è stata messa in dubbio a causa di molti programmi di elevato profilo in cui i risultati comportamentali dei roditori predicevano effetti analgesici assenti nell’uomo. Per esempio, è stato trovato che gli inibitori FAAH erano anti-nocicettivi in un gruppo di modelli animali, ma composti come PF-04457845 non producevano effetti analgesici in pazienti osteoartritici nonostante abbassassero di > 96% l’attività FAAH. Allo stesso modo, è stato visto che gli antagonisti NK1 (sostanza P) invertivano robustamente le risposte nocicettive nei roditori nel contesto dell’infiammazione e del danno ai nervi, ma non sono stati in grado di produrre analgesia nei successivi studi clinici. Nonostante ciò, molti analgesici usati clinicamente, come i FANS e gli oppioidi, producono effetti anti-nocicettivi nei roditori sebbene tipicamente a dosi molto più alte di quelle usate nei pazienti.
Sfruttare i modelli di dolore in modelli di organismi per rilevare possibili analgesici pone davanti a numerose sfide: 1) come misurare il dolore, un rapporto cosciente soggettivo di una esperienza sensoriale spiacevole, dal momento che non abbiamo accesso a ciò che gli animali sentono? 2) I modelli sono dei veri surrogati delle condizioni/malattie che normalmente producono dolore nei pazienti? 3) Bisogna vincere la sfida tecnica di come eliminare le distorsioni che confondono, le modificazioni indotte dall’osservatore e la mancanza di riproducibilità; e 4) i farmaci che hanno come bersaglio proteine umane possono non essere attivi sui loro omologhi roditori. La prima è la più difficile dal momento che possiamo misurare solo i risultati che possono essere correlati con alcuni aspetti del dolore, come la sottrazione allo stimolo o l’apprendimento di evitamento di una situazione che potrebbe essere dolorosa. Per le misure riflesse di dolore tipicamente uno stimolo breve che dura alcuni secondi si applica ad un’area del corpo dell’animale e viene misurata la risposta. Ciò porta chiaramente ad una scarsa corrispondenza con il dolore spontaneo in corso che è il problema maggiore dei pazienti. Sono stati esperiti tentativi di sviluppare misurazioni del risultato che possano riflettere la presenza del disagio ma questi richiedono un maggiore sforzo e validazione per renderli robusti ed utili. Proprio perché alcune classi di analgesici come gli oppiacei possono ridurre a dosi elevate i riflessi nocicettivi non significa che tali riflessi abbiano validità predittiva per tutte le classi di analgesici. Abbiamo bisogno di misurazioni che si avvicinano il più possibile ai livelli persistenti di disagio presenti negli animali; e non siamo ancora vicini.
Alcuni surrogati di malattia, come il dolore in risposta ad uno stimolo nocivo acuto, incisione di una ferita, o danno traumatico ad un nervo, sono facili da imitare. Molto più difficili sono i modelli di dolore cronico diffuso che fotocopino esattamente la malattia umana. Infine, una maggiore difficoltà con i modelli preclinici è che l’osservatore umano può introdurre modificazioni nel comportamento animale che possono distorcere la misurazione del risultato attraverso l’induzione di paura ed ansia. Recentemente, è stato mostrato che perfino il genere dell’investigatore ha un impatto significativo nel comportamento dei topi correlato al dolore. Migliori pratiche di laboratorio possono eliminare le distorsioni in quanto accurati studi in cieco e la ripetizione indipendente dovrebbe essere uno standard. In conclusione, mentre i modelli preclinici sono inestimabili nella validazione di bersagli e nelle misurazioni dell’azione farmacologica, attualmente non hanno valore predittivo e necessitano di una interpretazione cauta. Nuove tecnologie che usano misure quantitative del comportamento alterato in animali che si comportano liberamente per periodi prolungati in maniera indipendente dall’osservatore potrebbero trasformare l’utilità dei modelli preclinici nel prossimo futuro.
_______________________________________________________________________________________________

Figura 1. Meccanismi rilevanti di nocicezione ed analgesia
Ci sono molti bersagli sia nei neuroni periferici, che nelle sinapsi nocicettive tra glia, neuroni PNS e CNS nelle corna dorsali del midollo, che forniscono strategie di intervento per lo sviluppo di analgesici. La figura riassume alcuni dei principali bersagli che sono approfonditi nel testo.
Neurobiologia del dolore
I meccanismi che sostengono e guidano il dolore cronico (Fig. 1) sono già stati affrontati da altri. Il dolore è nelle situazioni fisiologiche una risposta protettiva necessaria iniziata da neuroni sensitivi periferici ad alta soglia in seguito al rischio di danno dovuti a stimoli o infezioni tessutali. Infatti, la presenza di dolore agisce come boa interna per allertare il corpo alla presenza di uno stimolo nocivo in modo che le necessarie risposte correttive possano essere messe in atto. Per danno e infezioni tissutali, l’ipersensibilità al dolore accompagna l’infiammazione associata e persiste per tutta la durata della risposta infiammatoria, aiutando il decorso della riparazione ed evitando l’uso della parte del corpo colpita. Tuttavia, il dolore può anche essere un maladattamento patologico quando insorge a causa di disfunzioni del sistema nocicettivo, a causa di fattori genetici, per esempio il disordine di dolore parossistico estremo dovuto a mutazioni di Nav 1.7 con acquisto di funzione, il danno del sistema nervoso centrale che porta a dolore neuropatico e a sindrome da amplificazione centrale abnorme. In tutti questi casi il dolore non è più un sintomo ma la malattia stessa.
Un’altra guida di dolore spontaneo in assenza di stimolazione periferica è l’attività ectopica di neuroni sensitivi primari (Fig. 1). Molti canali del sodio dipendenti dal voltaggio (Nav 1.3, Nav 1.7 e Nav 1.8), canali del potassio (KCNQ), canali del calcio (Cav 2.2) e canali attivati dall’iperpolarizzazione e modulati da nucleotidi ciclici (HCN) sono stati implicati nella modulazione dell’attività ectopica e della eccitabilità di membrana in neuroni sensitivi soprattutto dopo un danno dei nervi periferici. Tuttavia, deve essere ancora provata una relazione diretta di questa attività ectopica di neuroni sensitivi primari con il dolore spontaneo, a causa principalmente dei risultati inconsistenti e della scarsità di modelli preclinici affidabili, anche se l’imaging di cellule vive con attività reporter geneticamente codificata dovrebbe cambiare questa realtà.
Una conseguenza principale dell’infiammazione periferica è l’iperfunzione della trasduzione nocicettiva dei canali ionici nei terminali periferici dei neuroni sensitivi. Questi canali sono responsabili della conversione di stimoli nocivi in correnti centripete. La sensibilizzazione periferica di questi trasduttori e dei canali ionici coinvolti nella eccitabilità di membrana porta ad una riduzione nella soglia di attivazione e ad ipereccitabilità dei nocicettori nei neuroni sensitivi. Tuttavia, la sensibilizzazione periferica da sola non è completamente responsabile della prolungata presenza di dolore, dell’allodinia tattile dinamica, della sommazione temporale del dolore e della diffusione dell’ipersensibilità al dolore a tessuti non danneggiati (iperalgesia secondaria). Queste caratteristiche della ipersensibilità al dolore sono la conseguenza di un aumentato segnale sensitivo nel CNS dovuto ad un prolungato aumento dell’eccitabilità e dell’efficacia sinaptica di neuroni nocicettivi centrali e della perdita di attività inibitoria. Questo fenomeno, chiamato sensibilizzazione centrale, è comune a tutti i tipi di dolore clinico, alcuni dei quali sono guidati dall’attività dei nocicettori sotto forma di plasticità dipendente dall’attività, ed altri da modificazioni autonome nel sistema nervoso centrale.
Molti meccanismi contribuiscono alla sensibilizzazione centrale; aumentata forza sinaptica dovuta ad un aumento del numero dei canali ionici presinaptici, aumentato rilascio del neurotrasmettitore presinaptico o aumentata attività del recettore post-sinaptico del neurotrasmettitore. Anche la disinibizione, in cui la trasmissione inibitoria noradrenergica, oppioidergica e GABAergica è ridotta attraverso varie modificazioni sinaptiche e perdita degli interneuroni spinali gioca un ruolo principale, soprattutto nel dolore neuropatico. Inoltre, il danno al sistema nervoso potrebbe produrre modificazioni strutturali nei circuiti nocicettivi che portano ad aumentata sensibilità al dolore. A seconda della causa responsabile del dolore, molti dei processi descritti prima potrebbero anche agire di concerto, complicando ulteriormente le opzioni di trattamento.
In aggiunta ai meccanismi prima riportati, ci sono molte influenze modulatorie del dolore locali e distribuite. Gli endocannabinoidi, per esempio, stimolano i recettori cannabinoidi sia nelle sinapsi GABAergiche che glutammatergiche dove servono da trasmettitori retrogradi e bloccano la trasmissione di segnali del dolore. In aggiunta, i peptidi oppioidi endogeni come le endorfine, encefaline, dinorfina ed endomorfine stimolano diversi sottotipi di recettori oppioidi (mu, kappa e delta) per garantire analgesia in vari contesti.
I bersagli del sistema oppioide
I ligandi oppioidi esogeni isolati dai semi d’oppio del papavero sono stati usati come potenti analgesici per più di 3000 anni e oppioidi come idrocodone e morfina rimangono ancora gli analgesici di scelta per molti pazienti in clinica. Tuttavia, mentre stimolare i recettori oppioidi può portare ad una efficace inibizione del dolore soprattutto in un danno acuto o in uno scenario palliativo, i recettori oppioidi possono anche indurre effetti deleteri, come tolleranza, dipendenza fisica e un potenziale d’abuso che hanno portato ad una crisi nelle prescrizioni e all’abuso ricreazionale degli oppioidi. Sono quindi stati saggiati molti approcci per mitigare gli effetti deleteri degli analgesici oppioidi.
Oppioidi deterrenti dell’abuso
Molte compagnie si sono focalizzate nel modificare gli oppioidi esistenti per sviluppare formulazioni deterrenti dell’abuso (ADF). Per esempio, l’ossicodone a rilascio esteso (ER) (OxyContin, Purdue Pharma) e Opana ER (Oxymorphone, Endo Pharmaceuticals) sono comunemente prescritti in base all’assunzione che l’abuso avvenga solo con farmaci che hanno una cinetica rapida, che aumenta l’euforia. Tuttavia, ciò potrebbe essere non corretto. Mentre i dati clinici sono limitati e per lo più non conclusivi, alcuni dati mostrano che l’incidenza dell’abuso potrebbe addirittura aumentare con gli oppioidi ADF. Infatti, anche se formulati diversamente, gli agenti attivi sono comunque oppioidi euforizzanti ed i farmaci mantengono tutti gli effetti collaterali potenziali di questa classe, inclusi il potenziale di overdose in seguito ad esposizione a dosi elevate. In aggiunta, mentre OxyContin ADF ha ridotto il numero di prescrizioni per l’uso e l’abuso di OxyContin, molti tossicodipendenti hanno o imparato ad interferire con la deterrenza all’abuso o si sono spostati verso la prescrizione di altri oppioidi, fentanil o eroina.
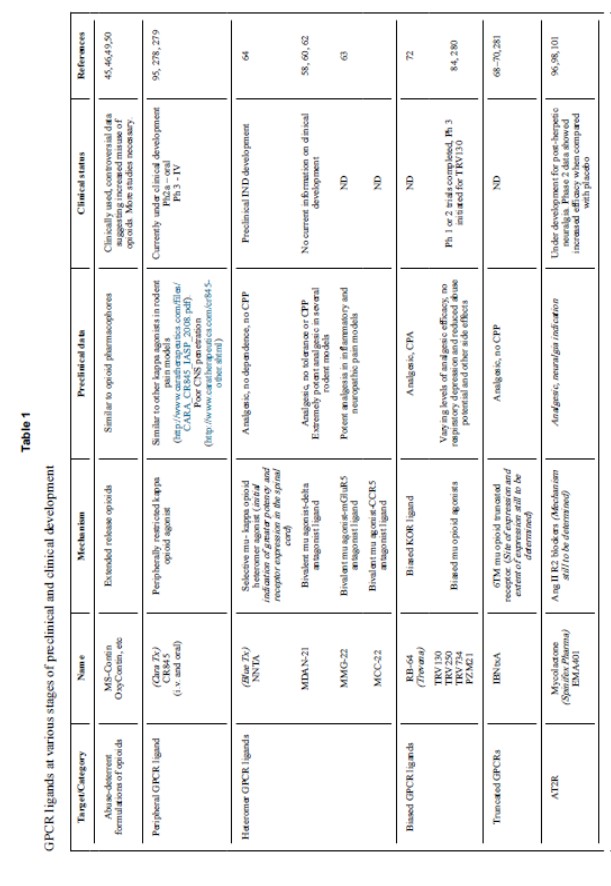
Ligandi kappa-selettivi ristretti perifericamente
Tutti i principali oppioidi analgesici attualmente nel mercato sono agonisti del recettore oppioide mu. Sono stati fatti notevoli sforzi per sviluppare ligandi selettivi delta e kappa dal momento che alcuni dei composti selettivi iniziali hanno mostrato una minore propensione alla dipendenza fisica e all’abuso. Infatti, molti agonisti kappa producono un comportamento avversivo lieve o grave. Tuttavia, questi sforzi non hanno avuto successo a causa o dell’analgesia inefficace (delta) o dei gravi effetti psicotomimetici e disforici (kappa). Per esempio, i ligandi kappa come Enadoline e butorfanolo sono stati sviluppati per avere un minore potenziale d’abuso, ma mentre danno ancora gratificazione, inducono però anche stanchezza, sedazione, confusione, ansia, distorsioni visive e uditive e spersonalizzazione.
Dal momento che molti degli effetti collaterali degli agonisti kappa sono generati nel CNS, Cara Therapeutics sta attualmente sviluppando un agonista kappa CR845 ristretto perifericamente per somministrazione sia IV che orale. E’ stato mostrato che CR845 IV inibisce con successo il dolore addominale post-operatorio in studi di Fase II e si sta attualmente saggiando una formulazione orale in pazienti con dolore cronico del ginocchio in uno studio di Fase II.
Questi dati clinici preliminari sono promettenti, ma rimane ancora aperto il problema dell’efficacia dei ligandi ristretti perifericamente nel ridurre la maggior parte delle forme di dolore cronico, soprattutto quelle sostenute da sensibilizzazione centrale, come già descritto.
Eteromeri dei recettori oppioidi, ligandi bifunzionali ed isoforme
Un altro approccio per eliminare gli effetti collaterali associati agli oppioidi mu si basa sulla rapida crescita di evidenze che i GPCR, inclusi i recettori oppioidi, formano sia oligomeri che complessi di ordine elevato (Fig. 2). Gli omodimeri (complessi di recettori simili) e gli eterodimeri (complessi di recettori diversi) forniscono dei nuovi bersagli potenzialmente interessanti per interventi farmacologici.
Sono stati generati molti ligandi bivalenti che contengono distinti farmacofori per due recettori legati insieme con linker ad atomi di carbonio di varia lunghezza che dimostrano profili farmacologici promettenti (Tabella 1). Per esempio, MDAN-21 (agonista μ/antagonista δ), una serie di ligandi agonisti mu/antagonisti CB1, MMG22 (agonista μ/antagonista mGluR5) e MCC22 (agonista μ/antagonista CCR5) hanno dimostrato una potente attività antinocicettiva in vari modelli preclinici di dolore nei roditori ed inoltre non possiedono tolleranza o preferenza di posto, una misura della tendenza all’abuso. Tuttavia, queste molecole sono piuttosto grandi, sollevando interrogativi sulla loro biodisponibilità. C’è quindi interesse nello sviluppo di piccole molecole come ligandi di recettori oppioidi eteromerici. NNTA, un eterodimero agonista selettivo del recettore oppioide mu-kappa, è un esempio. E’ stato trovato che i recettori oppioidi eterodimerici mu-kappa formano eterodimeri nel midollo spinale dei ratti e la presenza di questi recettori sembra più alta nei ratti femmina che non nei maschi. NNTA ha come bersaglio eterodimeri mu-kappa a concentrazioni sub-picomolari ed è un potente agente anti-nocicettivo che sembra nei roditori non provocare tolleranza, dipendenza fisica e comportamento di ricerca del farmaco. NNTA è attualmente sotto investigazione da parte di Blue Therapeutics attraverso studi IND-enabling per la sicurezza con piani futuri di studi clinici nell’uomo dopo aver riempito con successo il pacchetto IND. In aggiunta, ligandi bifunzionali come la piccola molecola BU08028, analogo della Buprenorfina, descritta da poco, per i recettori oppioidi mu e nocicettina, è un altro potenziale candidato farmaco che ha mostrato una potente anti-nocicezione senza depressione respiratoria o dipendenza fisica nelle scimmie.
Un approccio diverso, ma complementare si è focalizzato nel puntare a isoforme differenti del recettore oppioide mu (Tabella 1). Il gene orpm1 ha due promoter indipendenti e può quindi formare diverse varianti di splicing, tra cui alcuni recettori con domini 6TM inaspettati che sono strutturalmente alquanto diversi dai GPCR a domini 7TM. Per esempio, la variante di splice MOR-1C risulta in un recettore 6TM funzionalmente attivo e un ligando (IBNTxA) che stimola questo recettore produce anti-nocicezione senza comportamento di ricerca del farmaco in studi di condizionamento del posto di preferenza o depressione respiratoria nei topi. Ciò suggerisce che il dominio transmembrana C-terminale del recettore oppioide mu potrebbe non essere necessario per l’attracco di certi ligandi. Allo stesso modo, la variante di splice funzionale MOR-1D modificata all’estremità c-terminale media la sua dimerizzazione con GRPR per generare un oppioide indotto dal grattamento nei topi, fornendo una ulteriore evidenza che l’estremità c-terminale di MOR potrebbe giocare un ruolo nel produrre gli effetti avversi degli oppioidi. Molti dubbi tuttavia rimangono su questi segnali di recettori troncati, sulle dimensioni e livello di espressione tissutale, ed il perché stimolarli alteri gli effetti collaterali dell’oppioide mu. In aggiunta, ulteriori ricerche dovranno garantire la similarità tra le isoforme dei recettori oppioidi murini ed umani.
Ligandi distorsivi
Mentre è stato inizialmente considerato che i GPCR segnalano solo attraverso proteine G specifiche con cui sono accoppiati, la recente scoperta di ligandi distorsivi ha aumentato la nostra comprensione del segnale di trasduzione GPCR ed ha fornito nuove conoscenze sulla farmacologia GPCR. GPCR accopiati a β-arrestine possono essere stimolati o evitati da ligandi che restringono il recettore a conformazioni particolari, e possono quindi portare a distinti effetti farmacologici di uno stesso recettore (Fig. 2).
Sono stati sviluppati molti agonisti distorsivi di recettori oppioidi che producono effetti collaterali oppioidei ridotti con anti-nocicezione simile o migliore, almeno in modelli animali preclinici (Tabella 1). Un esempio cospicuo è TRV130, un agonista distorsivo del recettore oppioide mu che attiva il segnale della proteina G, ma non della β-arrestina. Studi clinici con TRV130 sono stati largamente positivi, ed hanno mostrato che è ben tollerato ed ha una efficacia analgesica comparabile alla morfina ma con un inizio più veloce. TRV130 produce anche depressione respiratoria, nausea e vomito significativamente ridotte nei pazienti, tuttavia ciò sembra essere dose-dipendente. Il fatto che la depressione respiratoria non è completamente eliminata con questo approccio è preoccupante, dal momento che la tendenza all’abuso di questo composto non è stato ancora determinato nell’uomo.
La recente eccitante scoperta di PZM21 – un agonista oppioide mu Gi distorsivo – illustra chiaramente che una farmacologia differenziale può essere ottenuta attivando selettivamente messaggeri di segnale specifici attraverso lo stesso recettore. Il composto è il prodotto di una estesa vagliatura assistita in silico di > di 3 milioni di ligandi ed è senza i comuni effetti collaterali degli oppioidi di costipazione, depressione respiratoria e comportamento di ricerca del farmaco. Tuttavia, rimangono alcuni problemi dal momento è stato trovato che l’efficacia analgesica della molecola è quattro volte meno potente della morfina in studi sui roditori.
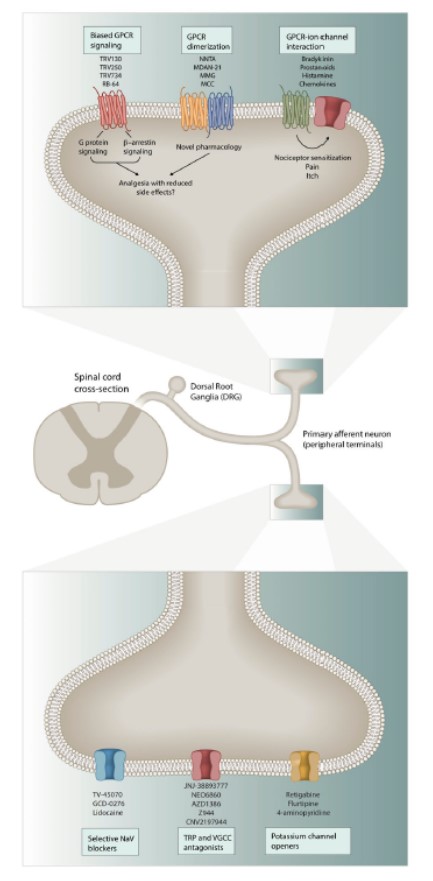
Figura 2. Bersagli per la scoperta di farmaci analgesici periferici
I terminali periferici di neuroni nocicettivi afferenti primari esprimono GPCR e vari canali ionici che mediano la trasduzione di diverse modalità sensitive, alcune delle quali hanno caratteristiche di accoppiamenti e segnali specifici che offrono nuove opportunità per nuove classi di farmaci. I GPCR possono utilizzare sia proteine-G che β-arrestine come secondi messaggeri e ligandi interferenti possono stimolare particolari segnali per aumentare l’efficacia e ridurre gli effetti avversi. I GPCR possono anche complessarsi fisicamente con altri recettori portando alla formazione di omodimeri ed eterodimeri e anche interagire funzionalmente con canali ionici come TRPV1, TRPA1, GIRK, etc. Sono stati anche sviluppati anticorpi anti-NGF (nerve growth factor) con l’intento di alleviare certe modalità di dolore bloccando l’attivazione di TrkA.
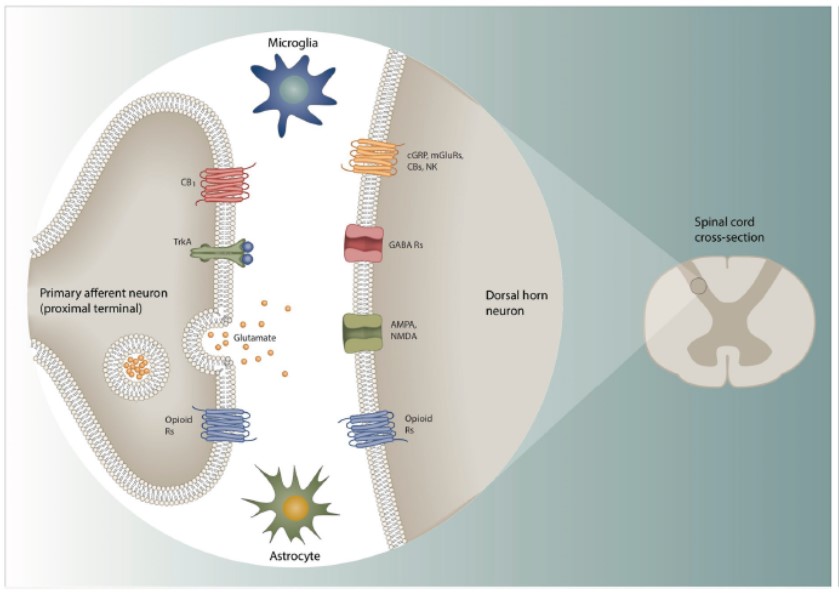
Figura 3. Avere come bersaglio il segnale tra i nocicettori pre-sinaptici ed i neuroni post-sinaptici nelle corna dorsali del midollo spinale
I terminali centrali dei neuroni sensitivi nocicettivi fanno sinapsi con neuroni delle corna posteriori del midollo spinale che dopo elaborazione da parte dei circuiti locali trasmettono le informazioni al cervello. I terminali nocicettivi pre-sinaptici forniscono input generati da stimoli nocivi nella periferia, infiammazione e danno ai nervi periferici ai neuroni post-sinaptici. Molti dei meccanismi di trasmissione/modulazione coinvolti rappresentano bersagli analgesici potenziali, o riducendo la trasmissione eccitatoria o aumentando quella inibitoria. Alcuni attori fondamentali – oppioidi, cannabinoidi, NK1 ed altri GPCR, insieme a canali ionici come NMDA, AMPA, e GABA sono discussi nell’articolo. Un altro attore fondamentale è il recettore TrkA che è legato da NGF – ed è stato visto che bloccandolo si produce una potente analgesia. Tuttavia, la mancanza di specificità delle vie nocicettive in molti casi rende gli effetti indesiderati da bersaglio problematici.
Canali ionici come bersagli farmacologici
I canali ionici sottendono tutte le attività elettriche dei neuroni. Di principio, mirare canali ionici per alterare il segnale del dolore potrebbe essere fatto a qualsiasi stadio, dall’inibizione della scarica dei nocicettori primari all’inibizione dei neuroni di secondo ordini o di ordini più elevati nel midollo spinale e nel cervello. La via più ovvia di mirare selettivamente al segnale del dolore è di puntare canali ionici che hanno una espressione preferenziale in nocicettori primari. Questa è la strategia incarnata nei farmaci che mirano ai canali TRPV1, che sono fortemente espressi nei nocicettori primari ed hanno una scarsa o nulla espressione in molti altri tipi neuronali. Anche molti altri potenziali bersagli farmacologici, inclusi i canali del sodio dipendenti dal voltaggio Nav1.8 e Nav1.9, sono altamente espressi nei nocicettori primari con scarsa espressione in una più ampia varietà di neurono sensitivi e simpatici periferici ma hanno scarsa espressione nel sistema nervoso centrale.
Mentre le strategie attuali si sono focalizzate su canali di neuroni sensitivi primari, sta anche emergendo il potenziale di mirare canali ionici in neuroni di ordine più alto. In questo caso, è probabilmente difficile o impossibile confinare gli effetti dei farmaci solo nelle vie nocicettive. Tuttavia, questo non è necessariamente un problema insormontabile. Anche molte terapie esistenti basate su canali ionici per altre condizioni mirano a canali con ampia espressione nel sistema nervoso, incluse farmaci anti-epilettici che mirano diffusamente a canali del sodio e farmaci ansiolitici che mirano a canali GABAA. In generale, le nostre conoscenze sull’organizzazione di neuroni e circuiti neuronali sono troppo primitive per capire come questi farmaci che si legano a canali ionici ampiamente espressi possano avere efficacia clinica senza effetti collaterali debilitanti. Appena raccoglieremo conoscenze più dettagliate sugli schemi di espressione, sulla regolazione e funzione di vari canali in neuroni di ordine più elevato nella segnalazione del dolore, sarà forse possibile puntare all’attività di questi neuroni in maniera razionale.
Canali TRPV1. Il clonaggio molecolare dei canali TRPV1 e la loro identificazione nel 1997 come recettori in neuroni sensitivi primari che mediano il dolore termico (Fig. 3) ha portato una valanga di studi che implicavano l’attivazione di TRPV1 in una varietà di modelli preclinici di dolore. I programmi di sviluppo dei farmaci che mirano i canali TRPV1 sono seguiti velocemente, con brevetti archiviati per oltre 1000 composti da circa 50 compagnie. Tuttavia, i risultati sono stati deludenti. In generale, l’efficacia degli antagonisti TRPV1 in condizioni di dolore clinicamente rilevanti nell’uomo è stata modesta, e ci sono due effetti avversi da bersaglio problematici visti con molti di questi composti: ridotta sensibilità al calore nocivo, che potenzialmente può portare a ustione accidentale, e un aumento della temperatura corporea, che riflette probabilmente la partecipazione dei canali TRPV1 nella normale regolazione della temperatura. Questi problemi, insieme alla modesta efficacia, hanno determinato una interruzione di molti programmi.
Tuttavia, un lavoro recente suggerisce che forse è possibile sviluppare composti che mantengono attività anti-nocicettiva evitando effetti avversi, trovando agenti che possono inibire l’attivazione aumentando la temperatura o abbassando il pH (che è stato suggerito possa mediare il controllo della temperatura corporea). Tali “antagonisti a modalità specifica” si basano sull’evidenza che ligandi vanilloidi come la capsaicina (e forse agenti endogeni che mediano il dolore) attivano TRPV1 in maniera diversa rispetto a temperatura e pH e che queste modalità di attivazione possono essere inibite differenzialmente. NEO6860 (Neomed) è uno di questi agenti a modalità specifica ed è attualmente in studio di Fase II per l’osteoartrite. Una nuova tecnologia che abilita strutture ad alta risoluzione dei canali TRPV1 legati ai ligandi usando la microscopia a crio-elettroni dovrebbe grandemente facilitare ulteriori sviluppi di questo approccio.
Canali del sodio. Canali del sodio dipendenti dal voltaggio sottendono segnali elettrici in tutti i neuroni. Ci sono nove tipi distinti di canali del sodio codificati da geni diversi, di cui tre (Nav1.1, Nav1.2, e Nav1.6) sono ampiamente espressi sia nel CNS che nel PNS (Fig. 2 e 3) e altri tre (Nav1.7, Nav1.8, e Nav1.9) sono espressi principalmente in sottotipi di neuroni periferici. I bloccanti dei canali del sodio attualmente usati, incluse lidocaina, amitriptilina, e mexiletina, mostrano una selettività relativamente piccola tra diversi canali del sodio, e la loro finestra terapeutica è limitata da effetti avversi indesiderati, principalmente da azioni sul CNS. Ciò ha stimolato sforzi per lo sviluppo di composti selettivi per i canali del sodio con scarsa espressione nel CNS.
Nav1.7 – I canali Nav1.7 sono espressi in nocicettori e neuroni simpatici, con solo una bassa espressione nel CNS. Sono emersi come candidati primari per nuovi analgesici in seguito all’identificazione di rare mutazioni umane in cui la perdita di funzione nei canali Nav1.7 è risultata in insensibilità congenita al dolore. In aggiunta, sono state identificate un certo numero di mutazioni Nav1.7 con acquisizione di funzione, in cui i pazienti provano dolore, qualche volta grave, o spontaneamente o innescato da stimoli normalmente innocui. Allo stesso modo, modelli di topi knockout per Nav1.7 mostrano un quasi completa insensibilità a stimoli nocicettivi e mancanza di dolore infiammatorio, mentre rispondono normalmente ad una varietà di stimoli non dolorifici.
Nav1.7 è attualmente attivamente studiato dall’industria farmaceutica. Sono state trovate piccole potenti molecole inibitrici dei canali Nav1.7, tra le più notevoli una classe di composti sulfonamidi che possono essere altamente selettivi per i canali Nav1.7 tra gli altri canali del sodio, e perfino selettivi per i canali Nav1.7 umani tra quelli di altre specie. Studi clinici di molti di questi composti sono in corso (Tabella 2), incluso il composto della Icagen/Pfizer PF-05089771 che ha completato studi di Fase II per la rimozione del dente del giudizio e per l’eritromialgia primitiva, e due composti (GDG-276 e GDC-0310) sviluppati da Xenon Pharmaceuticals che si stanno saggiando in studi di Fase I in collaborazione con Genentech.
Il sito di legame degli inibitori sulfonamidi è stato recentemente determinato ed è completamente differente da quello di piccole molecole inibitrici dei canali del sodio classiche come la lidocaina, carbamazepina, ed altri anestetici locali ed agenti anti-epilettici, che interagiscono con la regione poro dei canali del Na e generalmente hanno scarsa selettività tra i diversi sottotipi di canali del sodio. L’elevata specificità dei composti sulfonamidi e le conoscenze inusualmente dettagliate del suo sito di legame dovrebbero consentire il disegno di piccole molecole inibitrici di Nav1.7 perfino più potenti e selettive.
Si stanno saggiando anche molte classi di composti non-sulfonamidi che mirano primariamente i canali Nav1.7, ma sono generalmente meno selettivi. Raxatrigine (Convergence/Biogen) è in studio di Fase II per la neuralgia trigeminale, e funamide (TV-45070, Xenon/Teva) è in studio di Fase IIb per la neuralgia post-herpetica.
In aggiunta, sono stati sviluppati peptidi inibitori dei canali Nav1.7 da peptidi di origine naturale di veleno di ragno intelligentemente modificati. Questo approccio sfrutta le differenze in amminoacidi tra diversi tipi di canali del sodio in circuiti extracellulari delle proteine canale ed ha portato a peptidi inibitori con affinità molto elevata per il blocco dei canali Nav1.7 e ad un elevato grado di selettività dei sottotipi. Tuttavia, il rilascio di questi grandi composti peptidici potrebbe rappresentare una sfida significativa nel trasferimento alla clinica.
Un potenziale problema con l’uso clinico di bloccanti dei Nav1.7 è che mentre i canali sono minimamente espressi nel CNS, sono espressi nei neuroni olfattivi primitivi, il chè significa che un potente bloccante Nav1.7 potrebbe produrre anosmia, un effetto collaterale trovato intollerabile da una frazione consistente di pazienti negli studi clinici su farmaci con altri bersagli.
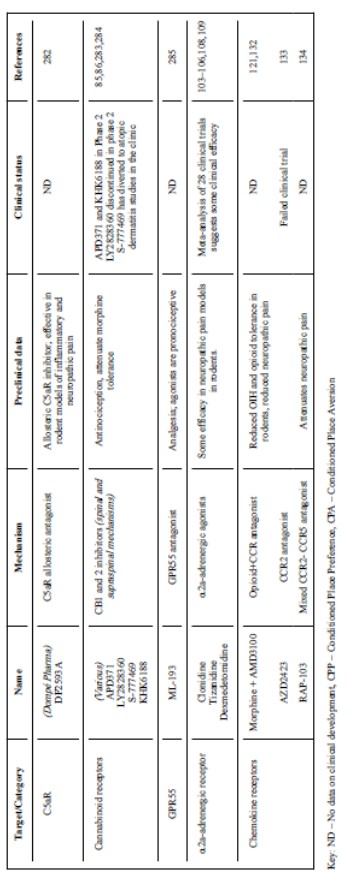
Un altro potenziale problema correlato alla recente scoperta che la sensibilità al dolore può essere restaurata dal naloxone sia nei topi senza Nav1.7 che in uomini senza Nav1.7. Ciò solleva la possibilità che la perdita di sensazione del dolore in mutanti senza Nav1.7 è più complicata della semplice rimozione funzionale dei canali
Nav1.7 e potrebbe risultare da un aumento di espressione delle vie oppioidee di inibizione del dolore. Tuttavia, è possibile che questo aumento rifletta complessi eventi compensatori durante lo sviluppo che non verrebbero mimati dall’inibizione acuta.
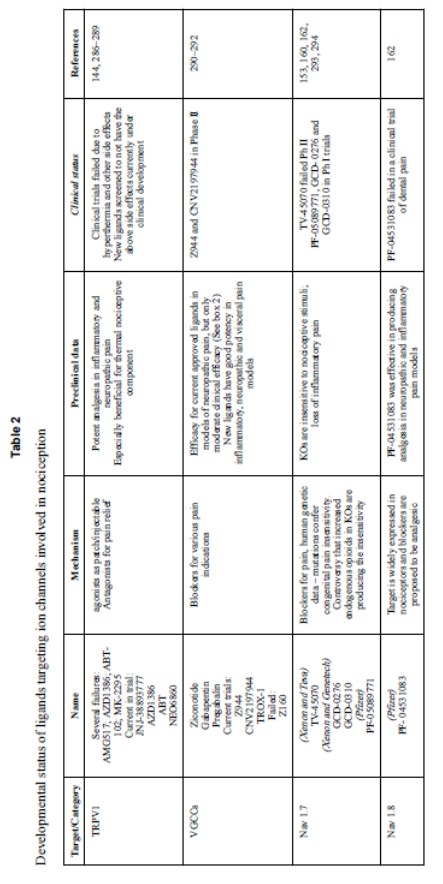
Nav1.8 – I canali Nav1.8 sono canali del sodio resistenti alla tetrodotossina e aperti dal voltaggio che sono espressi in neuroni nocicettivi primari di piccolo diametro, con assente o scarsa espressione in altri tipi neuronali. Di principio, la loro espressione selettiva ed il loro contributo dominante al totale delle correnti di sodio in neuroni DRG di piccolo diametro (almeno nel soma) fanno dei canali Nav1.8 dei bersagli farmacologici attraenti. Sono stati sviluppati inibitori Nav1.8 abbastanza selettivi ed hanno mostrato proprietà anti-nocicettive in modelli di roditori. Sono stati compiuti sforzi per sviluppare inibitori Nav1.8 per l’uso clinico nell’uomo. Tuttavia, il solo saggio clinico riportato di un inibitore selettivo Nav1.8, il composto Pfizer PF-04531083, è risultato in uno studio terminato sul dolore dentale e non ci sono altre evidenze. Nonostante i canali Nav1.8 sono preminenti nel corpo cellulare di neuroni nocicettivi DRG, la propagazione assonale in questi neuroni dipende generalmente da canali sensibili alla TTX. In un modello di ratto di dolore neuropatico, l’espressione dei Nav1.8 negli assoni è sufficientemente aumentata per consentire la propagazione resistente alla TTX di potenziali d’azione in alcune fibre C, ma rimane non chiarito in quali condizioni l’inibizione Nav1.8 possa inibire la generazione o la propagazione di potenziali d’azione nelle condizioni di dolore nell’uomo.
Nav1.9 – Come i canali Nav1.8, i canali Nav1.9 sono espressi in neuroni nocicettivi primari di piccolo diametro. I canali Nav1.9 si attivano ed inattivano molto lentamente ed in normali condizioni fisiologiche sembrano essere per lo più inattivati. La loro normale funzione fisiologica potrebbe essere quella di fornire una piccola corrente per aiutare a depolarizzare il potenziale di riposo ed amplificare le depolarizzazioni lente sottosoglia tra il potenziale di riposo e il picco di soglia. Topi geneticamente senza Nav1.9 mostrano una normale sensibilità al dolore acuto ma ridotta iperalgesia in risposta all’infiammazione, suggerendo che questi canali possano rappresentare bersagli terapeutici. Tuttavia non è evidente se Nav1.9 sia attualmente clinicamente utile. Una vagliatura per bloccanti di Nav1.9 è stata ostacolata dall’incapacità di esprimere Nav1.9 in sistemi eterologhi anche se recentemente è stato riportato un canale chimerico Nav1.9/Nav1.4 esprimibile.
Rilascio mirato di bloccanti dei canali del sodio carichi – Per inibire selettivamente l’attività elettrica di neuroni nocicettivi primari che innescano il dolore, una strategia alternativa per mirare i canali Nav1.7 o Nav1.8 è di usare canali a grandi pori selettivamente espressi da neuroni come portali per il rilascio di farmaci (Fig. 2). Molti nocicettori primari esprimono sia i canali TRPV1 o TRPA1 che entrambi. Questi canali sono insoliti in quanto formano un poro che permette l’ingresso di cationi molto grandi, incluso N-metil-D-glucamina (PM 195 Dalton) ed il colorante cationico FM1-43 (PM 452 Dalton). Questa proprietà può essere sfruttata per il rilascio di molecole farmacologiche cationiche dentro i neuroni. Il derivato della lidocaina QX-314 (N-etil-lidocaina) è una molecola cationica (PM 263 Dalton) che è inefficace nell’inibire i canali del sodio neuronali quando si presenta nel lato extracellulare del canale, perché è troppo grande per penetrare all’interno del poro del canale del sodio dall’esterno, ma può entrare ed inibire i canali quando si presenta dal lato intracellulare. Tale entrata avviene solo quando i canali sono aperti e la molecola QX-314 può essere intrappolata all’interno dei canali quando i canali si richiudono, così il blocco si accumula in maniera dipendente dall’uso. I pori dei canali TRPV1 e TRPA1 sono abbastanza grandi Perché QX-314 permei la cellula, con entrata della molecola cationica guidata non solo dal gradiente di concentrazione ma anche dal voltaggio di membrana intracellulare negativo, promuovendo concentrazioni più alte all’interno della cellula. Quindi, QX-314 applicato esternamente può entrare ed inibire i canali del sodio in neuroni dove i canali TRPV1 e TRPA1 sono attivati. Anche alcuni canali della famiglia P2X, inclusi P2X2, P2X4, e P2X7 hanno grandi pori e possono probabilmente ammettere QX-314 e molecole simili.
In esperimenti iniziali che studiano l’applicazione perineurale ai nervi sciatici in animali normali, la QX-314 co-applicata con la capsaicina ha prodotto una inibizione di lunga durata del segnale dolorifico con scarsi effetti sulla funzione motoria mediata dallo stesso tronco nervoso. In esperimenti successivi, QX-314 è stato co-applicato con lidocaina, che agisce da agonista sia dei canali TRPV1 che TRPA1 a concentrazioni mM. La lidocaina blocca tutte le funzioni dei nervi solo transitoriamente (circa 1 ora), mentre la co-applicazione con QX-314 ha prodotto un blocco nocicettivo che dura fino a 9 ore o più, il chè probabilmente riflette la lunga residenza di QX-314 una volta entrato nelle fibre che contengono TRPV1 e TRPA1.
In molte situazioni cliniche, i canali TRPV1 e TRPA1 potrebbero essere sufficientemente attivati endogenamente che non c’è bisogno di applicare un attivatore esogeno dei canali TRP insieme al bloccante cationico dei canali del sodio. Per esempio, durante il prurito, i canali TRPV1 e TRPA1 sono attivati da secondi messaggeri il cui rilascio è innescato da una varietà di recettori accoppiati a proteina G. L’attivazione di questi recettori è sufficiente per facilitare l’inibizione da QX-314 del prurito indotto sperimentalmente. Durante l’infiammazione, i canali TRPV1 e TRPA1 sono attivati da agenti endogeni, e in queste circostanze è probabile che sia non necessario co-applicare un attivatore esogeno dei canali TRP. Inoltre, poichè l’attivazione delle fibre-C e il rilascio di neuropeptidi fanno spesso parte del processo infiammatorio, inibire l’attività elettrica delle fibre-C può effettivamente ridurre l’infiammazione, non solo bloccare il dolore o il prurito, conseguenze dell’infiammazione. Il silenziamento delle fibre-C in seguito alla penetrazione di bloccanti caricati dei canali del sodio può ridurre l’infiammazione delle vie aeree in modelli animali di asma.
Una limitazione di questa strategia è che i bloccanti cationici dei canali del sodio devono essere applicati in stretta vicinanza alle fibre nervose da silenziare, dal momento che come molecole cationiche hanno una limitata penetrazione tissutale. Metodi di applicazione adatti includono l’instillazione diretta nelle ferite, iniezioni locali, e rilascio tramite aerosol per influenzare l’infiammazione polmonare. Anche l’applicazione nella cute danneggiata nelle dermatiti o in condizioni simili potrebbe essere efficace. E’ degno di nota che questa limitata penetrazione tissutale significa che c’è una limitata o nulla esposizione sistemica agli agenti cationici, e questo minimizza grandemente le possibilità di effetti indesiderati.
Canali del calcio. I canali del calcio dipendenti dal voltaggio sono canali selettivi per il calcio che sono chiusi con potenziali di riposo normali e si aprono con la depolarizzazione. Sottindendono a varie funzioni neuronali incluso l’aiuto a regolare l’eccitabilità cellulare e a mediare il rilascio di vescicole sinaptiche. Su dieci canali del calcio dipendenti dal voltaggio codificati dal genoma di mammifero, otto hanno una diffusa espressione nei neuroni.
Gli agenti terapeutici che attualmente sono usati più diffusamente e che hanno come bersaglio i canali del calcio dipendenti dal voltaggio sono gabapentina (Neurontin) e pregabalin (Lyrica), che si pensa agiscano influenzando il traffico ed il riciclo delle proteine dei canali del calcio. Per trattare iperalgesia e dolore neuropatico, gabapentina e pregabalin sono efficaci solo in una minoranza di pazienti, e non si sa ancora il perché. E’ stato proposto che la migliorata selettività terapeutica può essere ottenuta da agenti selettivi per le subunità alpha2delta-1, dal momento che queste subunità sembra medino gli effetti anti-iperalgesici di gabapentina e pregabalin, mentre i farmaci legano anche le subunità alpha2delta-2, che potrebbero mediare gli effetti collaterali. In aggiunta, avanzamenti recenti nella nostra comprensione di un paio di canali del calcio hanno aperto alcune altre strade nel disegno di farmaci analgesici.
Varianti di splice dei Cav2.2 – La trasmissione sinaptica è innescata dall’entrata del calcio attraverso i canali del calcio presinaptici. La maggior parte della trasmissione sinaptica nel cervello di mammifero e nel midollo spinale è mediata da canali del calcio Cav2.1 (del tipo P/Q) e Cav2.2 (tipo N). E’ interessante che la trasmissione nelle prime sinapsi delle vie del dolore, le sinapsi glutammatergiche dai neuroni sensitivi primari ai neuroni di secondo ordine nelle corna dorsali, sembra che contino quasi esclusivamente sui canali presinaptici Cav2.2. I topi knock-out per Cav2.2 hanno una ridotta sensibilità ad alcuni ma non a tutti i tipi di dolore acuto, infiammatorio, e neuropatico. I canali Cav2.2 possono essere bloccati selettivamente da diversi peptidi del veleno delle lumache cono e uno di questi, ω-conotossina MVIIA (ziconitide, Prialt), è attualmente usata per trattare il dolore, con applicazione per mezzo di pompa intratecale. Tuttavia, la sua utilità è limitata dalla piccola finestra terapeutica, che probabilmente riflette la partecipazione dei canali Cav2.2 in altre funzioni del midollo spinale e forse l’eccesso del peptide in regioni più alte del sistema nervoso.
L’efficacia di Prialt ha stimolato sforzi per trovare piccole molecole inibitrici dei canali Cav2.2 che potrebbero essere efficaci contro il dolore. Sono stati riportati molti di questi inibitori, tra cui TROX-1. Come piccole molecole inibitrici dei canali del sodio, questi sono inibitori dipendenti dallo stato, che si legano più strettamente agli stati attivati del canale che a stati vicini al riposo. Quindi, l’inibizione dipendente dallo stato risulta in una inibizione maggiore dei canali che stanno ciclando ripetitivamente e velocemente attraverso stati attivati, conferendo potenzialmente una certa selettività per neuroni che sono iperattivi. Nonostante molti di questi inibitori di canali del calcio dipendenti dallo stato hanno dimostrato efficacia nel liberare dal dolore in molti modelli di roditori, l’efficacia in studi sull’uomo deve ancora essere dimostrata.
Dal momento che i canali Cav2.2 sono ampiamente espressi nel cervello, un bloccante del canale disponibile sistemicamente che attraversi la barriera emato-encefalica dovrebbe probabilmente esercitare seri effetti indesiderati. Tuttavia, i canali Cav2.2 in neuroni sensitivi primari hanno diverse varianti di splice rispetto ai canali predominanti nel cervello, e ciò solleva la possibilità di sviluppare piccole molecole con una sufficiente selettività per ridurre la trasmissione nel midollo spinale senza effetti centrali deleteri.
Cav3.2 – I canali Cav3.2 sono canali di tipo-T a bassa soglia che in vari neuroni centrali possono mediare il picco di scarica in seguito ad iperpolarizzazione (che rimuove l’inattivazione da riposo dei canali). La loro normale funzione fisiologica nei neuroni sensitivi è enigmatica, ma eliminare o diminuire l’espressione dei canali Cav3.2 con RNA anti-senso ha un effetto analgesico notevole nel dolore meccanico e termico dei ratti, sollevando la possibilità che possano essere bersagli terapeutici. Il composto ABT-639 della Abbvie, un inibitore Cav3.2 disponibile oralmente con una ragionevole selettività ha prodotto anti-nocicezione dose-dipendente in un modello nel ratto di dolore alle articolazioni del ginocchio ed ha ridotto l’allodinia tattile in molti modelli nei roditori di dolore neuropatico, inclusi la legatura dei nervi spinali e la CCI. Tuttavia, studi clinici successivi su ABT-639 con volontari umani non hanno mostrato effetto nel modello di dolore acuto da capsaicina intradermica in cui pregabalin era efficace. Allo stesso modo, ABT-639 non ha ridotto il dolore in pazienti con neuropatia diabetica. Altri inibitori Cav3.2 sono stati descritti come efficaci in modelli di dolore, incluso un modello nel ratto di dolore da sindrome dell’intestino irritabile.
Molte molecole che hanno attività bloccante dipendente dallo stato sia su canali Cav2.2 che 3.2 (e potenza variabile nel bloccare altri canali del calcio come i Cav2.1) sono stati descritti ed è stato riportato che sono efficaci in modelli nel ratto di dolore neuropatico, dolore infiammatorio, ed allodinia meccanica. Tuttavia, non è chiaro se composti correlati verranno studiati per un possibile uso clinico.
Canali del Potassio. I neuroni di mammifero esprimono molti tipi diversi di canali del potassio, con schemi di espressione significativamente diversi tra vari tipi di neuroni. L’espressione di un certo numero di canali del potassio diversi è diminuita in neuroni sensitivi primari di modelli animali di iperalgesia e dolore neuropatico, Il chè probabilmente contribuisce alla conseguente ipereccitabilità di neuroni che segnalano il dolore. Di principio, correnti in aumento attraverso i canali del potassio potrebbero essere una via efficace di inibire l’attività neruronale, e gli schemi di espressione differenziale di vari tipi di canali del potassio offrono la possibilità di inibire l’attività di una specifica popolazione neuronale, come i nocicettori, senza una eccessiva alterazione di altre funzioni neuronali. La ricerca di aumentatori delle correnti di potassio per inibire il segnale del dolore è relativamente recente, ma è già evidente che la strategia generale potrebbe essere promettente. Ad oggi, la maggiore attenzione per questo approccio è stato sulla famiglia di canali Kv7 (KCNQ), e molti membri della famiglia di canali a “due-pori” (K2P). Un approccio alternativo per piccole molecole stimolanti la funzione dei canali è provare a correggere i cambiamenti dell’espressione genica dei canali del potassio associati a dolore.
Canali Kv7 – I canali Kv7 sono canali dipendenti dal voltaggio che tipicamente si attivano e deattivano in maniera relativamente lenta e spesso possono essere attivati da piccole depolarizzazioni sottosoglia. La descrizione originale di una corrente neuronale Kv7 è stata “corrente-M”, chiamata così per la sua capacità di essere inibita dalla stimolazione muscarinica in neuroni simpatici e successivamente è stato mostrato che è mediata da canali eteromerici Kv7.2/Kv7.3. Attraverso una lenta attivazione con piccole depolarizzazioni, la corrente Kv7 produce un forte adattamento di scarica evocato da depolarizzazione di stato. Sembra che i canali Kv7.2/Kv7.3 siano ampiamente espressi nel sistema nervoso, spesso con una forte espressione nel segmento assonale iniziale, dove sono ben posizionati per modulare le azioni dei potenziali di scarica. Sono stati trovati un certo numero di composti per aumentare l’attivazione dei canali della famiglia Kv7 facendo slittare la dipendenza dal voltaggio verso voltaggi più negativi. Il meglio conosciuto tra questi è retigabina, introdotto nell’uso come farmaco anti-epilettico. Retigabina inibisce i comportamenti nocicettivi in modelli di dolore neuropatico ed iperalgesia nel roditore. Se gli effetti siano mediati da neuroni centrali o periferici non è chiaro. Con disappunto, tuttavia, uno studio clinico sul dolore neuropatico nell’uomo non ha raggiunto il risultato primario di efficacia, il chè aggiunge la retigabina alla lunga lista di composti con efficacia nei modelli di dolore dei roditori che sembrano avere effetti molto meno drammatici nei pazienti umani.
Un altro composto che aumenta la corrente mediata da canali Kv7 spostando l’attivazione verso voltaggi più negativi è flupirtina, che appartiene alla stessa classe chimica della retigabina (differendo solo per la sostituzione di un anello piridinico con un anello fenile). È interessante notare che la flupirtina è stata introdotta nella pratica clinica in Europa nel 1984 come analgesico non oppioide che agisce a livello centrale. Gli effetti della flupirtina potrebbero essere mediati sia centralmente che perifericamente. Neuroni DRG di piccolo diametro probabilmente corrispondenti a fibre-C esprimono subunità Kv7.2 ed una componente della loro corrente di potassio è inibita dal bloccante Kv7 XE991. Eliminazione genica dei Kv7.2 in tutti i neuroni sensitivi somatici produce un modesto aumento di sottrazione da stimoli dolorosi meccanici e termici, mediato principalmente dalla mancanza di Kv7.2 dalle fibre A-delta, forse riflettendo l’espressione preminente di canali Kv2 nei nodi di Ranvier di molti nervi mielinizzati. Di particolare interesse è che la subunità primaria Kv7 espressa dalle fibre-C nocicettive di piccolo diametro è Kv7.5, il chè solleva la possibilità che lo scrutinio di stimolatori selettivi di questa subunità possano identificare nuovi analgesici che agiscono perifericamente. Tuttavia, Kv7.5 è preminentemente espresso anche nella muscolatura liscia vascolare, così che questi agenti potrebbero anche diminuire la pressione sanguigna. Tuttavia, se i canali delle fibre-C nocicettive sono monomeri Kv7.5 e quelli della muscolatura vascolare sono eteromeri con altre subunità Kv7, ci potrebbe essere un’opportunità come bersaglio selettivo.
Canali K2P – La famiglia di canali a due pori (K2P) costituisce un gruppo diverso di 15 prodotti genici con ciascuna proteina che contiene quattro segmenti transmembrana e due domini a pori, con canali formati dai dimeri delle subunità. I canali K2P sono ampiamente espressi sia in neuroni che in cellule non neuronali. Molti canali K2P sono costitutivamente aperti, e i canali K2P sottolineano la principale conduttanza di potassio a riposo responsabile per il potenziale di riposo negativo delle cellule. A causa della loro regolazione sia del potenziale di riposo che della conduttanza in entrata dei neuroni, i canali K2P possono esercitare un forte controllo sull’eccitabilità neuronale. Per questa ragione, i composti che aumentano l’apertura dei canali K2P (o forse aumentano la loro espressione) sono una possibilità attraente per lo sviluppo di farmaci. In paragone a molti altri canali, si sa relativamente poco sul contributo funzionale di canali specifici K2P in vari tipi neuronali, molto perché ci sono attualmente pochi strumenti farmacologici specifici. Nonostante ciò, scoperte recenti suggeriscono che molti canali K2P potrebbero essere bersagli clinici attraenti per alleviare il dolore. L’espressione di un certo numero di canali K2P espressi in neuroni DRG è diminuita dopo danno al nervo e in vari altri modelli animali di dolore neuropatico associato ad ipersensibilità neuronale. Di particolare interesse per il suo ruolo nei nocicettori è TREK1, che si basa sulla scoperta sorprendente che TREK1 può essere attivato dalla morfina attraverso i recettori oppioidi mu, con riduzione di circa metà dell’azione analgesica della morfina in animali con TREK1 geneticamente eliminato. Attualmente, si sa troppo poco sull’espressione relativa di vari canali K2P in diversi tipi neuronali (e non neuronali) per predire quale potrebbe essere il bersaglio migliore per alleviare il dolore, minimizzando al contempo disturbi ad altre funzioni. Lo sviluppo di strumenti farmacologici specifici, che è in atto in molti lab, dovrebbe essere un passo chiave.
Canali Kv2/Kv9 – I canali Kv2 sono attivati dal voltaggio e rettificano in maniera ritardata le correnti del potassio e sono ampiamente espressi sia nei neuroni centrali che periferici. Le subunità Kv2.1 presentano in modo particolare una espressione ampia ed elevata in molti tipi neuronali. Di particolare interesse per lo sviluppo di nuovi farmaci, le subunità Kv2 possono formare canali eteromerici con subunità di altre famiglie geniche, tra cui le subunità Kv5, Kv6, Kv8, e Kv9, chiamate subunità “silenti” che non sembrano formare canali funzionali da sole. Nel contesto del dolore, l’attenzione si è incentrata in modo particolare sulle subunità Kv9.1, per cui una variane allelica comune umana è fortemente associata con aumentata suscettibilità al dolore neuropatico. Successivi studi animali hanno mostrato che l’espressione delle subunità Kv9.1 è fortemente diminuita in neuroni sensitivi primari dopo danno ai nervi, in parallelo allo sviluppo di comportamenti dolorifici. E’ stato ipotizzato che la riduzione dell’espressione di Kv9.1 possa produrre scarica spontanea nelle fibre A-delta. Quindi, una strategia farmacologicamente plausibile sarebbe quella di trovare piccole molecole stimolanti dei canali che contengono Kv2.1, o puntando i rimanenti canali Kv2.1/Kv9.1 o a qualsiasi combinazione eteromerica sia favorita dopo l’espressione diminuita dei canali Kv9.1. Anche i canali Kv2.1 sono fortemente espressi in neuroni DRG con fibre-C di piccolo diametro, dove sono potenzialmente associati con una varietà di altre subunità silenti, incluse Kv6.1, Kv8.1, Kv9.2, e Kv9.3. Le potenzialità dei canali basati su Kv2 costituiti da combinazioni altamente diversificate di subunità in diversi tipi neuronali, sollevano la possibilità di trovare piccole molecole con selettività differenziale per particolari combinazioni di subunità che potrebbero ridurre preferenzialmente l’eccitabilità di neuroni che segnalano il dolore. La possibilità di selettività farmacologica che punta diverse combinazioni di subunità è validata dalla sensibilità differenziale dell’inibitore ad ampio spettro 4-aminopiridina, ma ciò non è stato ancora esplorato sistematicamente per la funzione di stimolazione.
Canali ionici come bersagli in neuroni di ordine più elevato
Nel dolore neuropatico, c’è molto probabilmente una alterata eccitabilità e alterati circuiti di neuroni nocicettivi di ordine più elevato nel midollo spinale e nel cervello. Si sa poco sui canali ionici coinvolti nel controllo dei cambiamenti di eccitabilità nei neuroni di ordine più alto, ma, con l’aumento della comprensione, sarà forse possibile puntare selettivamente questa attività. Agenti che riducono l’attività dei canali del sodio ampiamente senza selettività di sottotipo, come l’amitriptilina, potrebbero agire bene nell’alleviare il dolore modificando l’attività di neuroni di ordine più alto così come di neuroni sensitivi primari (Fig. 3).
La discussione sul meccanismo d’azione dei gabapentinoidi si è generalmente incentrata sulla trasmissione sinaptica nelle sinapsi di primo ordine nel midollo spinale, dal momento che gli effetti sul traffico nei canali del calcio è stato studiato nei neuroni DRG, ma questi effetti potrebbero anche essere presenti nelle sinapsi di ordine più alto. Modulatori allosterici positivi dei canali del cloro GABAA potrebbero avere delle potenzialità nell’alleviare il dolore, ma generalmente producono anche una forte sedazione. E’ stato recentemente trovato che flupirtina agisce aumentando le correnti mediate da GABAA a concentrazioni paragonabili a quelle che aumentano l’attività Kv7. Poiché i canali GABAA sono altamente diversi in termini di subunità strutturali (essendo combinazioni pentameriche di 19 diverse potenziali subunità) con differenti combinazioni in diversi neuroni e regioni sub-cellulari, il chè è ancora non capito in modo molto completo, è possibile trovare agenti con selettività per specifiche combinazioni di subunità che aumentano le proprietà anti-dolorifiche in paragone alla sedazione. Una migliore comprensione degli schemi di espressione di combinazioni specifiche di subunità del recettore GABAA nei circuiti neuronali di ordine più alto che mediano il dolore sarebbe utile per un approccio razionale in questa direzione.
Enzimi come bersagli farmacologici analgesici
I FANS, gli analgesici usati più ampiamente, hanno come bersaglio l’enzima ciclossigenasi e c’è un interesse crescente nell’individuare di altri bersagli enzimatici nello sviluppo di nuovi analgesici (Tabella 3).
Sintesi BH4 – reduttasi sepiapterin. Il danno assonale aumenta la produzione di tetraidrobiopterina (BH4) dal GTP in neuroni danneggiati, un cofattore essenziale per l’idrossilasi di amine aromatiche, per le sintasi dell’ossido nitrico e la monossigenasi alchilglicerolo, a causa dell’aumentata espressione di GTP cicloidrolasi 1 (GCH1), l’enzima limitante nella via BH4 nelle cellule danneggiate. Il danno al nervo è anche accompagnato da un aumento della sintesi di BH4 nei macrofagi che si infiltrano in prossimità del danno. Studi genetici nell’uomo rivelano un’associazione tra polimorfismi GCH1 che diminuiscono i livelli di BH4, e riduzione del dolore nei pazienti, e diminuzione dei livelli di BH4 tramite inibizione o eliminazione genica di GCH1 in neuroni sensitivi produce analgesia nei roditori. L’inibizione di GCH1 tuttavia crea il rischio di eventi avversi nei sistemi nervoso e cardiovascolare a causa della diminuita sintesi di amine biogeniche e NO se BH4 scende eccessivamente al di sotto dei livelli basali. Una strategia alternativa per minimizzare il rischio di effetti collaterali è avere come bersaglio la reduttasi sepiapterina (SPR), L’enzima terminale nella via sintetica di BH4, il cui blocco consente la produzione di abbastanza BH4 attraverso la via di salvataggio di BH4 per prevenire effetti avversi, mentre contemporaneamente si riducono i livelli elevati che contribuiscono al dolore neuropatico ed infiammatorio. L’inibizione di SPR produce analgesia in molteplici modelli di dolore acuto e cronico neuropatico ed infiammatorio senza tolleranza e senza effetto sulla nocicezione acuta. Una caratteristica attraente dell’inibizione di SPR per lo sviluppo di farmaci è che la stimolazione del bersaglio può essere determinata misurando i livelli di sepiapterina nel plasma e il biomarcatore di efficacia tramite riduzione dei livelli di BH4. E’ interessante che uno scrutinio tramite yeast three hybrid usando farmaci con attività clinica ma senza bersagli conosciuti ha portato alla scoperta che la sulfasalazina, un farmaco approvato dall’FDA usato per ridurre l’infiammazione ed il dolore ad essa associato nell’artrite reumatoide e nella malattia infiammatoria dell’intestino, è un inibitore SPR, anche se con potenza molto bassa. Quartet Medicine, attraverso una cooperazione strategica con Merck, sta sviluppando nuovi inibitori SPR per il trattamento del dolore infiammatorio e neuropatico.
Sintasi-1 della prostaglandina E2 microsomiale. La sintasi-1 della prostaglandina E2 microsomiale è responsabile della produzione di PGE2 durante l’infiammazione come enzima terminale nella via dei prostanoidi, e agisce a valle della ciclossigenasi 2. PGE2 è il principale sensibilizzatore infiammatorio dei nocicettori, ed è stato mostrato che l’inibizione di PGES1 usando MF-63 [2-(6-choloro-1H-phenanthro-[9,10-d]imidazol-2-yl)isophthalonitrile] e due nuovi inibitori 2-acilamminoimidazolo riducono la sintesi di PGE inducibile senza sopprimere la generazione di prostaciclina. I due nuovi inibitori di mPGES1 producono un’analgesia equivalente al diclofenac in un modello di porcellini d’india di dolore all’articolazione del ginocchio (modello di artrite mono iodoacetato). L’inibizione di mPGES1 rappresenta quindi un nuovo approccio per la gestione del dolore infiammatorio, probabilmente senza gli effetti avversi cardiovascolari (infarto miocardico) e di sanguinamento gastrointestinale degli inibitori della COX2.
Idrolasi eposside solubile. Gli acidi epossieicosatrienoici (EET) sono metaboliti derivati da P450-epossigenasi dell’acido arachidonico che agiscono come molecole endogene di segnale in molteplici sistemi biologici. L’idrolasi eposside solubile (sEH) è l’enzima che degrada gli EET, le cui azioni coinvolgono l’analgesia. Un inibitore di sEH, acido trans-4-[4-(3-trifluoromethoxyphenil-1-ureido)che -cyclohexyloxy]-benzoico (t-TUCB) stabilizza i mediatori bioattivi, acidi grassi epossi (EpFA) che sono scarsamente abbondanti, ma sono lipidi bioattivi altamente potenti che mantengono l’omeostasi ed hanno azione analgesica in un modello murino di neuropatia diabetica. Gli EpFA agiscono come modulatori a monte della via di stress ER, che si è visto è stimolata nella neuropatia diabetica e la cui riduzione porta ad analgesia. Tuttavia, dal momento che alcuni EET sensibilizzano i nocicettori, l’inibizione di sEH è probabile che aumenti il dolore infiammatorio. Questo probabilmente complicherebbe lo sviluppo di questi inibitori come analgesici, dal momento che i dolori infiammatorio e neuropatico potrebbero coesistere.

MECCANISMI EMERGENTI
Proteasi
Molte proteasi – proteasi a serina (tripsina), metalloproteasi della matrice (MMP), proteasi a cistina (catepsina S), caspasi e recettore PAR2 attivato da proteasi sono stati implicati nella modulazione del dolore. Livelli elevati di proteasi a serina e PAR2 sono stati osservati in topi con dolore da cancro, che è attenuato dall’inibizione dei livelli di proteasi o completamente eliminato in topi deficienti in PAR2. E’ stato mostrato che un aumento transitorio di MMP9 e un ritardato, ma persistente aumento dei livelli di MMP2 induce e mantiene il dolore neuropatico nei roditori. Lo stesso studio ha anche mostrato che gli inibitori MMP possono produrre anti-nocicezione, suggerendo che le MMP possono essere un bersaglio per lo sviluppo di terapeutici. Si dovrebbe notare tuttavia che le MMP sono anche coinvolte in molte altre vie ed inibitori ad ampio spettro possono causare effetti collaterali tossici. Infatti, studi clinici con inibitori di MMP per il cancro non hanno avuto successo a causa di tossicità e di mancanza di efficacia. Allo stesso modo, è stato mostrato che sia caspasi-6 che catepsina S regolano le interazioni neurone-glia durante il dolore neuropatico e potenzialmente portano a sensibilizzazione centrale.
L’espressione di serpinaA3N, un inibitore endogeno della proteasi a serina, è aumentata in neuroni sensitivi danneggiati dopo danno assonale e sembra che protegga contro il dolore neuropatico in quegli animali in cui l’aumento di espressione è maggiore. Topi che non hanno serpinaA3N sviluppano maggiore allodinia meccanica dopo danno ai nervi rispetto ai topi normali, e la somministrazione di SerpinaA3N esogena attenua l’ipersensibilità meccanica. La serpinaA3N inibisce l’elastasi leucocitaria (LE) che è rilasciata dai linfociti T che infiltrano i DRG dopo danno ai nervi. E’ stato mostrato che la perdita genetica di LE o un inibitore di LE (Sivelestat) attenua l’allodinia meccanica in modelli neuropatici. Queste scoperte rivelano un complesso interscambio tra i neuroni sensitivi danneggiati e le cellule immuni ed identificano un nuovo bersaglio cellulare immune per ulteriori valutazioni come analgesico nel dolore neuropatico.
SFIDE E CONSIDERAZIONI NELLO SVILUPPO CLINICO DI NUOVI ANALGESICI
La valutazione del fallimento delle prove di principio di efficacia in studi clinici su nuovi analgesici è circondata da rapporti insufficienti da parte dell’industria e, di conseguenza, è spesso incerta se sono selezionati il bersaglio, il farmaco, le misure dei risultati o i pazienti sbagliati, se manca la stimolazione del giusto bersaglio, o se è troppo forte una risposta placebo. Se non si impara niente da questi studi di fase II è cumulativamente uno spreco enorme di risorse ed alcuni farmaci possono essere abbandonati prematuramente.
E’ particolarmente preoccupante la possibilità che modelli preclinici non siano surrogati reali della malattia nell’uomo e che misure riflesse del “dolore” evocato da stimoli possano non essere predittori sensibili dell’efficacia nel paziente (Box 1). In molti casi, la manifestazione di effetti avversi significa semplicemente che non è possibile ottenere nell’uomo i livelli di esposizione al farmaco necessari per avere efficacia negli animali. In aggiunta, un grande effetto placebo e il controllo emotivo del dolore, tra le altre ragioni, complicano ulteriormente il disegno di studi clinici analgesici.
L’illustrazione delle sfide nel valutare l’azione di nuovi analgesici possono essere raccolte da alcuni recenti studi sul dolore che stanno esplorando nuovi bersagli di “dolore”:
Fallimento predittivo di modelli preclinici
I roditori, soprattutto i topi, rimangono l’organismo del modello preclinico di scelta per validare i bersagli e poi saggiarne i ligandi, usando tipicamente misure di comportamento correlati al dolore. Dal momento che molti meccanismi e vie neurobiologiche e di malattia sono evolutivamente conservati, molte informazioni utili possono essere raccolte utilizzando animali normali o geneticamente modificati. Tuttavia rimangono dei problemi importanti, innanzitutto c’è un dibattito considerevole sulla rilevanza delle risposte riflesse evocate a stimoli nocivi in condizioni di dolore clinico, specialmente il dolore spontaneo e le modificazioni della funzione cognitiva e dell’umore. Secondo, molti modelli di dolore non sono buoni surrogati della malattia nell’uomo (Box 1 e 2). Infiammare o danneggiare un’articolazione, per esempio, non ne fa un buon modello di osteoartrite. Sotto ci sono alcuni esempi dove l’efficacia clinica può non essere replicata in studi clinici nell’uomo.
Propentofillina – L’agente modulante la glia propentofillina non ha avuto successo in uno studio condotto da Solace Pharmaceuticals nel diminuire il dolore in pazienti con neuralgia post-herpetica, a dispetto di una certa attività in modelli di neuropatia periferica nei roditori. La spiegazione più probabile per questo fallimento potrebbe essere una importante differenza di specie tra roditori e uomo, sia nell’attivazione microgliale che nell’azione della propentofillina. Il coinvolgimento della microglia e degli astrociti nel dolore nell’uomo deve essere ancora ulteriormente saggiato ed il fallimento della propentofillina indica l’importanza sia della validazione dei bersagli nell’uomo, più comunemente con la genetica, sia dell’azione dei farmaci nelle cellule umane.
Inibizione della via p38 – l’inibitore di p38 MAP chinasi losmapimod non ha mostrato efficacia in pazienti con dolore neuropatico da radicolopatia lombosacrale, nonostante che studi preclinici avessero mostrato il coinvolgimento della via di segnale p38 nello sviluppo di dolore persistente dopo danno ai nervi periferici. Non è sicuro se lo studio non ha avuto successo per mancanza di stimolazione di un adeguato bersaglio o per differenze tra la radicolopatia lombosacrale cronica (anni) e modelli animali di dolore neuropatico molto più acuti (giorni/settimane). Poiché i dati preclinici non erano allineati con la coorte di pazienti saggiati, la significatività del mancato successo dello studio per altri tipi di dolore è incerta, il chè è vero per molti studi sul dolore neuropatico. Tuttavia, l’attivazione di p38 è a valle dell’NGF, un bersaglio validato per nuovi analgesici (vedi più sotto) e per l’angiotensina, che sembra agire in periferia per bloccare la sensibilizzazione di TRPV1. E’ degno di nota che un antagonista del recettore per angiotensina di tipo 2 (ATR2) EMA401 è stato efficace nella neuralgia post-herpetica, rappresentando il primo farmaco che agisce su un nuovo bersaglio e che mostra efficacia nel dolore neuropatico per un certo tempo. Quando è stato saggiato EMA401 il suo meccanismo analgesico d’azione era incerto. Tuttavia si sono dovuti abbandonare gli antagonisti TRPV1 a causa di ipertermia da bersaglio.
Ottenere la penetrazione nel CNS
Molti bersagli rilevanti per il dolore sono espressi nel sistema nervoso centrale (CNS). Tuttavia, la barriera emato-encefalica è una effettiva barriera o per la penetrazione nel CNS di ligandi di alcune strutture chimiche e per il raggiungimento dei loro bersagli o perché alcuni composti sono attivamente pompati fuori dal cervello perfino quando riescono ad entrare. Ciò è difficile da predire e può portare a insuccesso degli studi clinici.
L’endocannabinoide anandamide e il 2-arachidonoilglicerolo, agendo sui recettori CB, sono metabolizzati da enzimi idrolitici (idrolasi degli acidi grassi amidici (FAAH) e monoacilglicerolo lipasi (MGL)) e da enzimi ossigenanti (cicloossigenasi-2, COX-2). Un inibitore irreversibile dell’idrolasi-1 degli acidi grassi amidici PF-04457845, non è stato in grado di ridurre il dolore da osteoartrite del ginocchio, nonostante un decremento di > 96% dell’attività FAAH, sostanzialmente aumentando i substrati acidi grassi amidici endogeni, avendo molti studi preclinici mostrato efficacia in modelli di dolore infiammatorio. Nonostante questo insuccesso FAAH, altri composti stanno entrando in studi clinici e c’è un supporto genetico preliminare nell’uomo per il coinvolgimento di FAAH nella sensibilità al dolore da freddo. L’insuccesso del composto della Pfizer è da attribuire al mancato accesso FAAH nel CNS? Certamente, gli antagonisti cannabinoidi (CB1) hanno azioni principali nell’uomo tra cui viene reclamata un’azione sull’analgesia. L’uso clinico di agonisti CB1 o della cannabis è complicato dalla tendenza all’abuso farmacologico per gli effetti psicoattivi, da cui l’attrazione teorica per endocannabinoidi elevati.
Effetti avversi come conseguenza del coinvolgimento di meccanismi protettivi del dolore.
Mentre il dolore cronico può essere molto debilitante e ridurre la qualità di vita, In molte condizioni cliniche il dolore in corso protegge i tessuti danneggiati da ulteriori insulti. Per esempio, è stato provato che gli anticorpi anti-NGF (tanezumab, fulranumab e fasinumab) sono inusualmente più efficaci in pazienti con osteoartrite (OA) rispetto a ciò che si poteva prevedere in base ai dati preclinici, con segnali maggiori rispetto ai farmaci di riferimento, FANS e oppioidi, per il dolore cronico della parte bassa della schiena – che qualche volta si pensa sia la tomba degli studi analgesici – così come della OA e della neuropatia diabetica. Al contrario, è stato trovato che il tanezumab produce una rapida progressione dell’OA soprattutto quando viene somministrato a dosi elevate e con i FANS. Mentre la causa di ciò è incerta, una possibilità è l’eccessivo uso delle articolazioni danneggiate poiché l’elevata copertura analgesica potrebbe accelerare la distruzione articolare – il dolore è dopo tutto un meccanismo protettivo così come un problema patologico. Una preoccupazione aggiuntiva deriva dal ruolo di fattore di crescita dell’NGF nello sviluppo. Nelle scimmie cynomolgus gravide il tanezumab endovenoso aumenta la mortalità perinatale ed infantile, diminuisce la crescita infantile con modificazioni nel sistema nervoso periferico simpatico e sensitivo incluso lo sviluppo di neurotossicità. Questo rischio, così come la possibilità di una certa neurotossicità nell’adulto necessita di essere attentamente monitorato ed escluso.
Conclusioni – la strada da percorrere
Ci sono molti bersagli emergenti che sono promettenti per il trattamento del dolore acuto e cronico. Tuttavia, la domanda chiave è ancora se i dati di efficacia preclinica si possono tradurre nell’uomo, e se ciò non dovesse succedere, perché. Mentre l’high-throughput screening (HTS) in linee cellulari eterologhe genera ligandi selettivi, la mancanza di vie normali che influenzano il “bersaglio di interesse” nel suo ambiente cellulare originale potrebbe portare alla selezione di ligandi subottimali. In questo contesto, gli scrutini fenotipici compiuti direttamente su neuroni umani differenziati da linee iPSC di pazienti possono aiutare nell’aumentare fortemente la qualità dei dati rilevanti per i pazienti stessi. Un argomento simile si ha con molti dei modelli comunemente usati di evitamento riflesso nel comportamento doloroso nei roditori (Box 1). Dato che i roditori sono animali da preda, il loro comportamento può essere altamente influenzato dall’ambiente e dall’investigatore. Insieme, questi problemi suggeriscono che abbiamo bisogno di compiere uno sforzo significativo per sviluppare saggi che possono misurare automaticamente il dolore spontaneo, senza la presenza di alcun investigatore e il più vicino possibile a modelli di condizioni di malattie umane (Box 1).
Infine, bisogna focalizzarsi su bersagli che hanno già dati genetici nell’uomo che supportano nuovi analgesici efficaci rispetto a quelli che si basano solo su modelli di dolore nei roditori. Tuttavia, potrebbe essere un errore studiare solo bersagli con dati genetici nell’uomo, dal momento che molti bersagli dei farmaci più ampiamente usati clinicamente non sarebbero stati selezionati in base a varianti genetiche che producono chiari fenotipi correlati alla malattia. In aggiunta, grandi rari fenotipi come l’insensibilità al dolore potrebbero non essere utili ad identificare bersagli stimolati in condizioni cliniche, come dopo un danno al sistema nervoso o in risposta all’infiammazione cronica.
A dispetto di ciò, combinazioni di scrutini fenotipici in neuroni derivati da pazienti, in genetica umana, nell’editing genomico in topi e linee cellulari umane, così come miglioramenti nel disegno e nella cinetica dei farmaci, l’identificazione di biomarcatori e nel disegno di studi clinici (Box 2), tutti insieme offrono un’aumentata probabilità di successo, e ciò è qualcosa “dolorosamente” necessario. Dal momento che nuove opportunità terapeutiche derivano da questi sforzi, queste si aggiungeranno all’armamentario del dolore in mano ai medici creando opportunità per un approccio di medicina di precisione, dove i trattamenti possono essere mirati su meccanismi particolari che guidano il dolore presente in pazienti individuali.
Box 2
Disegno di studi clinici sul dolore
Studi clinici di fase 2 per prove di concetto su nuovi analgesici sono assediati da un certo numero di problemi principali; come misurare il dolore, come differenziare l’effetto placebo da quello farmacologico, e quale paziente selezionare? Il dolore è un’esperienza soggettiva influenzata non solo da tratti ereditari ma anche da processi psicosociali, cultura, esperienze, contesto sociale e biologico, e umore. Tradurre questa complessa esperienza dinamica interna in descrittori verbali, numerici, o scale analogiche di intensità, potrebbe non catturare livelli paragonabili di sensazione tra o all’interno dei soggetti, anche tramite istruzioni, e certamente non riflette il grado o la natura dell’iniziatore che precipita il dolore o del mantenitore del dolore. In aggiunta, l’alto livello dei sensibili al placebo e nocebo negli studi sul dolore cronico sono tra i principali fattori di confusione. Si stanno facendo tentativi di convivere con le distorsioni e l’inattendibilità, soprattutto con ACTTION, Analgesic, Anestethic, and Addiction Clinical Trial Translations, Innovations, Opportunities, and Network, una società pubblico-privato con la Food and Drug Administration (FDA) degli Stati Uniti. Il disegno degli studi può essere migliorato da una migliore diagnosi, studi in cieco e scelta delle misurazioni degli obiettivi primari, da disegni adattivi, incroci, e sottrazioni casuali, uso del potere adeguato e dell’analisi dei sensibili. Tuttavia, rimane il problema che la mancanza di biomarcatori predittivi del dolore o della stimolazione del bersaglio rende difficile capire la sottostante patofisiologia che porta a varie presentazioni cliniche del dolore e se il farmaco sta avendo qualsiasi azione, anche se le nuove tecnologie di imaging possono modificare questo aspetto. Mentre la ricerca preclinica sul dolore si è evoluta nel classificare vari tipi di dolore in base al meccanismo, poco di ciò è stato tradotto nella diagnosi di dolore in clinica. Studi recenti hanno proposto molte nuove vie di approcciare a questo problema: a) una metodologia consequenziale di classificazione può essere usata per identificare lo stato dolorifico, il meccanismo del dolore ed il bersaglio molecolare in sequenza – enfatizzando lo sviluppo di una terapia individualizzata o una medicina di precisione, b) identificare gruppi di pazienti clinicamente rilevanti in base ai meccanismi patofisiologici, e similmente c) accoppiare pazienti in base alle caratteristiche fenotipiche predittive di variazioni individuali. Questi sforzi sono un primo passo per classificare il dolore clinico in modo che si presti a migliorare il disegno degli studi clinici e degli interventi farmacologici significativi e dove l’efficacia possa essere prontamente identificata e quantizzata. Ciò sarà critico perché se un farmaco sta agendo su un bersaglio che gioca un ruolo solo in certi meccanismi che operano per produrre dolore, allora lo studio clinico avrà più successo se la presenza di questi meccanismi può essere identificata nei criteri di inclusione dei pazienti per arricchire quelli potenzialmente sensibili.
(Tratto da Ajay S Yekkirala, David P Roberson, Bruce P Bean, Clifford J Woolf, “Breaking barriers to novel analgesic drug development”, Nat Rev Drug Discov, 2017;16(8):545-564)
